Catalyse homogène — Chimie de coordination — Chimie Organométallique — Chimie du Palladium — Chimie de l’Or — Chimie du Gallium — Propriétés électroniques des ligands — Chimie du carbone divalent — Chimie des métaux nobles — Chimie des métaux du groupe principal — Chimie du Fer.
Catalyse au Fer
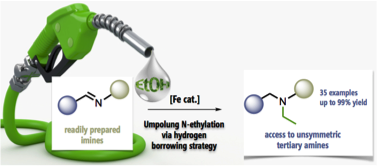
L’une des thématiques que j’ai initié en 2017, est basée sur la stratégie d’emprunt d’hydrogène pour la formation de nouvelles liaisons C‒N et C‒C. Tout d’abord la synthèse d’amines tertiaires éthylées dissymétriques ayant un large éventail d'applications en tant que produits pharmaceutiques et ou entrant dans la composition de matériaux ont focalisé notre attention. Les méthodes classiques d'éthylation des amines reposent sur l'utilisation de réactifs toxiques tels que l'acétaldéhyde et les halogénures d'éthyle. Pour rendre ce processus plus efficace et éco-compatible, nous avons développé une éthylation réductrice inédite d’imines catalysée par le fer, efficace et simple, en utilisant l'éthanol comme bloc de construction C2. Cette méthodologie est basée sur une stratégie d'emprunt d'hydrogène qui est une alternative aux méthodologies conventionnelles. Un complexe de fer stable à l'air est utilisé comme catalyseur et l'éthanol est à la fois le solvant et l'agent d'éthylation. Dans ces conditions, une variété d'imines portant des groupes aromatiques ou alkyle riches en électrons sur l'atome d'azote ont pu être efficacement alkylées par réduction, sans nécessiter l'utilisation d'hydrogène moléculaire. Le mécanisme de cette réaction, qui présente une sélectivité complète pour l'éthanol, a été étudié expérimentalement et au moyen de calculs DFT. Enfin, nous avons développé une méthode simple qui facilite la synthèse d'amines tertiaires éthylées non symétriques.
![]()
|
Angew. Chem. Int. Ed. 2018, 57, 3228 |
Plus récemment (2020) nous nous sommes intéressés au principe d’auto-transfert d’hydrogène, interrompu dans une réaction monotope de Wittig couplée à l’auto-transfert d’hydrogène. Nous sommes à présent capable à partir d’alcools primaires ou secondaires et d’ylures de phosphonium, de former de nombreux alcènes de configuration E. Sa version non interrompue vient d’être découverte, avec un catalyseur de manganèse muni d’un ligand PNP. Contrairement à la version interrompue, la réaction de Wittig est effectuée à̀ partir de sels de phosphonium et d’alcools primaires, pour former diverses briques moléculaires hydrogénées.
Les Anions Faiblement Coordinants et le Cation Vinylique
Basé sur nos travaux antérieurs nous avons souhaité étudier le comportement du contre-ion aluminate faiblement coordinant, [Al(OC(CF3)3)4] dans des processus catalytiques à base de métaux des cationiques comme les sels de gallium(I), d’indium à faible degré́ d'oxydation ainsi que des sels de lithium. Nous avons ainsi étudié́ les transformations catalysées par [Ga][Al(OC(CF3)3)4], notamment pour la dihydroarylation des arènes, l'hydrogénation par transfert des alcènes en utilisant le 1,4-cyclohexadiène comme source d'hydrogène, la cyclisation hydrogénante en tandem des arènes et la cycloisomérisation des énynes. Par la suite, nous avons démontré́ que le catalyseur [In][Al(OC(CF3)3)4] était très sélectif pour favoriser l'ortho-alkylations d'anilines non protégées en présence des styrènes.
 Ga
Ga
Par extension nous avons appliqué ce dernier à l'hydroamination d’alcénylamines primaires et secondaires non protégées dans des conditions douces. Enfin, le complexe [Li][Al(OC(CF3)3)4] s'est révélé́ être un catalyseur efficace pour la synthèse de dérivés du styrène à partir de vinyl triflate et d'arènes par l'intermédiaire d'un cation vinylique. Nous avons prouvé́ que cet anion volumineux et inerte [Al(OC(CF3)3)4] était capable d'apprivoiser des cations hautement réactifs que ce soient des métaux cationiques ou des intermédiaires réactionnels, comme le cation vinylique ouvrant ainsi de nouvelles perspectives en méthodologie de synthèse.
 Vinycation
Vinycation
Développement et Application de nouveaux complexes de Gallium et d’Indium
Nous avons développé de nouvelles familles de catalyseurs cationiques Ga(I), Ga(III), In(I) et In(III) capables d'imiter le comportement des complexes de métaux de transition nobles dans la catalyse des acides π ou l'hydrogénation par transfert. Ces espèces présentent des ligands NHC ou fluorobenzène. Elles se comportent comme des acides de Lewis doux capables de déclencher la formation de carbocations non classiques hautement délocalisées à partir d'alcynes et d'alcènes. Ils sont particulièrement utiles dans les réactions de réorganisation du squelette conduisant à des composés polycycliques après formation de liaisons C‒C ou C‒H. Plus récemment, nous avons développé plusieurs réactions monotopes mettant en jeu divers types d’activation. La première consiste à exploiter l’acidité de Lewis s et p du catalyseur [IPr·GaCl2][SbF6] pour promouvoir une réaction de métathèse carbonyl-ène, suivie d’une hydrogénation d’alcène par transfert d’hydrogène au moyen de 1,4-cyclohexadiène. Divers cyclopentanes et cyclohexanes ont été formés par cette méthode.
") Ga(III)
Ga(III)
Mécanismes réactionnels et développement de nouvelles réactions
Afin de prédire le modèle de réactivité général des acides π, et de distinguer entre deux métaux lorsqu'un mélange à deux composants est utilisé, ou si des protons adventices entrent en compétition comme véritable catalyseur, nous avons conçu un substrat contenant une fraction 7-alcynyl cycloheptatriène, qui combine la chimie des énynes et l'hydroarylation des allènes. Alors que les acides π-Lewis mous déclenchent un réarrangement de carbocation non classique pour générer deux indènes régioisomères A et B, les acides plus durs donnent plutôt le produit d'hydroarylation C ou le composé polycyclique D à la suite de deux étapes d'hydroarylation séquentielles.
Grâce à ce composé spécifique, nous sommes en mesure de classer l'acidité π-Lewis de divers systèmes catalytiques, de différencier le métal actif dans un mélange à deux composants, de détecter la présence de protons adventices et d'identifier les substituts de métaux nobles.
 tropyl
tropyl
Le test du cycloheptatriène a été utilisé avec certains nouveaux complexes cationiques NHC-Zn(II) et NHC-Al(III) (NHC=N-carbène hétérocyclique) ainsi que d'autres cations de zinc et d'aluminium dérivés de sels d'halogénure simples. Le premier s'est avéré capable d'agir comme un acide de Lewis π mous, ce qui a été confirmé par son activité dans une variété de transformations (hydrogénation par transfert, hydroalcoxylation, métathèse carbonyle-oléfine). L'avantage du ligand volumineux IPr‧NHC a été démontré par comparaison avec les sels simples de ZnX2. Le catalyseur NHC-Al(III) testé n'est pas capable d'activer les liaisons π C‒C mais de simples ions AlX2+ se sont révélés puissants dans certains cas.
 tropyltransfo
tropyltransfo
Enfin, la réactivité de 7-alkynycycloheptatriènes attachés à un groupe aryle sous catalyse π-acide a été étudiée. Une variété de produits cycliques utiles a été synthétisée via une réorganisation du squelette catalysée par Au(I), une hydroarylation catalysée par Cu(II) ou une réaction tandem hydroarylation/Friedel-Crafts catalysée par un acide de Brønsted. Nous rapportons également un type rare de réorganisation du squelette impliquant le 1,3-acétonide en présence d'un complexe Ga(I)+ cationique univalent.
Le N-bromosuccinimide a été utilisé comme agent de bromation pour la formation de bromoallenes trisubstitués à partir d'alkynylcycloheptratriènes. Divers phénylallènes portant une chaîne alkyle (avec ou sans oxygène sur la chaîne alkyle) ou un groupe silyle ont été synthétisés avec des rendements modérés à excellents. Enfin, la post-fonctionnalisation de la liaison C‒Br via des réactions de couplage croisé catalysées par Pd- ou Cu a donné différents allènes substitués.
Par la suite notre attention s’est portée sur l’étude de carbocations. De nombreux mécanismes réactionnels les font intervenir, en particulier des cations propargyliques et vinyliques. De nouvelles méthodologies de synthèse ont pu être développées, en impliquant ces deux familles de carbocations. Nous avons d’abord montré comment le motif bromoallène, dont le mécanisme de formation passe par un cation vinylique, pouvait être utilisé comme plateforme pour la première réaction de C—H propargylation d’arènes catalysée par un métal du groupe principal, le gallium. Par le biais d’études cinétiques, nous avons pu confirmer que l’étape cinétiquement déterminante du mécanisme de la réaction correspondait à l’abstraction du brome pour former un cation propargylique. Ces résultats ont été corroborés par la RMN du 71Ga et des études DFT qui ont mis en évidence la formation d'une paire d'ions à partir du bromoallène et du GaCl3 par abstraction de Br- (étape déterminant la vitesse). L'attaque du cation propargyle par l'arène et la déprotonation de l'intermédiaire de type Wheland fournissent alors le produit de propargylation C-H.
 CH propargyl
CH propargyl