Artificial photosynthesis
Axis 2 - CATALYTIC REDUCTION OF CO2 AND H2 PRODUCTION FOR ENERGY CONVERSION
Led by Zakaria Halime (zakaria.halime@universite-paris-saclay.fr)
Hinged Carboxylate in the Artificial Distal Pocket of an Iron Porphyrin Enhances CO2 Electroreduction at Low Overpotential. A. Smith, P. Gotico, R. Guillot, S. Le Gac, W. Leibl, A. Aukauloo, B. Boitrel,* M. Sircoglou,* Z. Halime,* Advanced Science (2025)
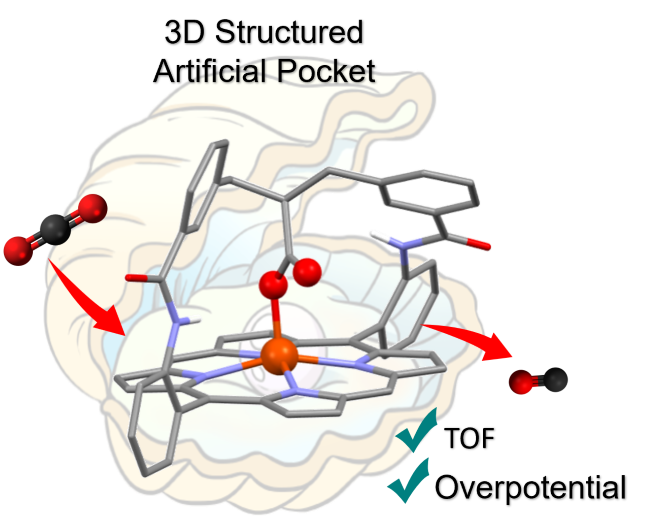
To efficiently capture, activate, and transform small molecules, metalloenzymes have evolved to integrate a well-organized pocket around the active metal center. Within this cavity, second coordination sphere functionalities are precisely positioned to optimize the rate, selectivity, and energy cost of catalytic reactions. Inspired by this strategy, an artificial distal pocket defined by a preorganized 3D strap was introduced on an iron-porphyrin catalyst (sc-Fe) for the CO2-to-CO electrocatalytic reduction. Combined electrochemical, kinetic, and computational studies demonstrate that the adequate positioning of a carboxylate/carboxylic group acting in synergy with a trapped water molecule within this distal pocket remarkably enhances the reaction turnover frequency (TOF) by four orders of magnitude compared to the perfluorinated iron-tetraphenylporphyrin catalyst (F20Fe) operating at a similar low overpotential. A proton-coupled electron transfer (PCET) was found to be the key process responsible for the unexpected protonation of the coordinating carboxylate, which, upon CO2 insertion, shifts from the first to the second coordination sphere to play a possible secondary role as a proton relay.
Time-Resolved Mechanistic Depiction of Photoinduced CO2 Reduction Catalysis on a Urea-Modified Iron Porphyrin. D. H. Cruz Neto, E. Pugliese, P. Gotico, A. Quaranta, W. Leibl, K. Steenkeste, D. Peláez, T. Pino,* Z. Halime,* M.-H. Ha-Thi,* (2024) Angew. Chem. Int. Ed., e202407723

The development of functional artificial photosynthetic devices relies on the understanding of mechanistic aspects involved in specialized photocatalysts. Modified iron porphyrins have long been explored as efficient catalysts for the light-induced reduction of carbon dioxide (CO2) towards solar fuels. In spite of the advancements in homogeneous catalysis, the development of the next generation of catalysts requires a complete understanding of the fundamental photoinduced processes taking place prior to and after activation of the substrate by the catalyst. In this work, we employ a state-of-the-art nanosecond optical transient absorption spectroscopic setup with a double excitation capability to induce charge accumulation and trigger the reduction of CO2 to carbon monoxide (CO). Our biomimetic system is composed of a urea-modified iron(III) tetraphenylporphyrin (UrFe(III)) catalyst, the prototypical [Ru(bpy)3]2+ (bpy=2,2’-bipyridine) used as a photosensitizer, and sodium ascorbate as an electron donor. Under inert atmosphere, we show that two electrons can be successively accumulated on the catalyst as the fates of the photogenerated UrFe(II) and UrFe(I) reduced species are tracked. In the presence of CO2, the catalytic cycle is kick-started providing further evidence on CO2 activation by the UrFe catalyst in its formal Fe(I) oxidation state.
Second Coordination Sphere Effect Shifts CO2 to CO Reduction by Iron Porphyrin from Fe0 to FeI. A. Sk,* P. Gotico, M. Sircoglou, W. Leibl, M. J. Llansola-Portoles, T. Tibiletti, A. Quaranta, Z. Halime,* A. Aukauloo* (2024) Angew. Chem. Int. Ed. Engl., e202314439
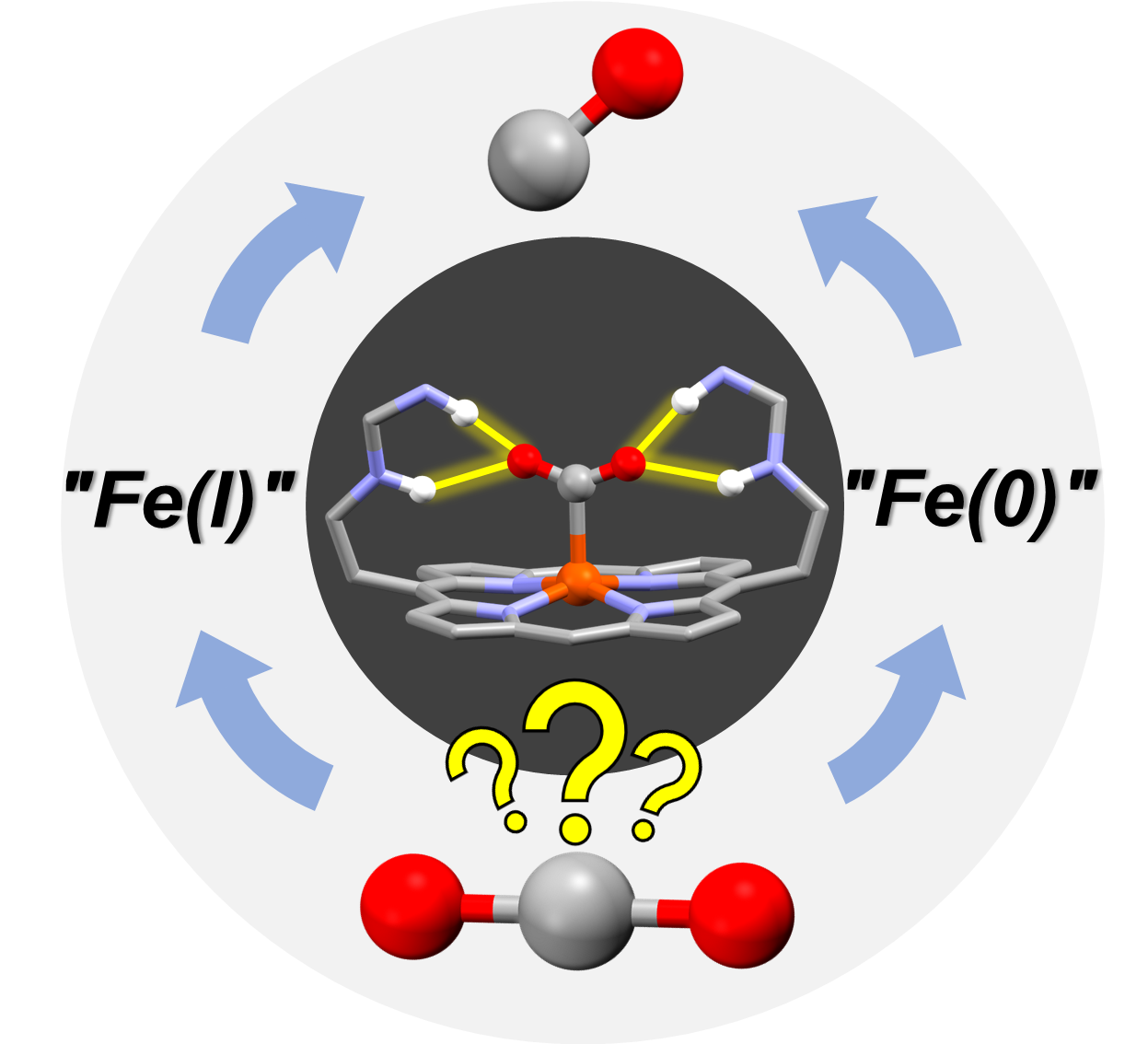
Iron porphyrins are among the most studied molecular catalysts for carbon dioxide (CO2) reduction and their reactivity is constantly being enhanced through the implementation of chemical functionalities in the second coordination sphere inspired by the active sites of enzymes. In this study, we were intrigued to observe that a multipoint hydrogen bonding scheme provided by embarked urea groups could also shift the redox activation step of CO2 from the well-admitted Fe(0) to the Fe(I) state. Using EPR, resonance Raman, IR and UV-Visible spectroscopies, we underpinned a two-electron activation step of CO2 starting from the Fe(I) oxidation state to form, after protonation, an Fe(III)‒COOH species. The addition of another electron and a proton to the latter species converged to the cleavage of a C‒O bond with the loss of water molecule resulting in an Fe(II)‒CO species. The DFT analyses of these postulated intermediates is in good agreement with our collected spectroscopic data, thus allowing us to propose an alternative pathway in the catalytic CO2 reduction with iron porphyrin catalyst. Such a remarkable shift opens new lines of research in the design of molecular catalysts to reach low overpotentials in performing multi-electronic CO2 reduction catalysis.
Unlocking Full and Fast Conversion in Photocatalytic Carbon Dioxide Reduction: Applications in Radio-Carbonylation. S. Monticelli, A. Talbot, P. Gotico, F. Caille, O. Loreau, A. Del Vecchio, A. Malandain, A. Sallustrau, W. Leibl, A. Aukauloo, F. Taran, Z. Halime,* D. Audisio* (2023) Nat. Commun. 14, 4451-2373
Harvesting sunlight to drive carbon dioxide (CO2) valorisation represents an ideal concept to support a sustainable and carbon-neutral economy. While the photochemical reduction of CO2 to carbon monoxide (CO) has emerged as a hot research topic, the full CO2
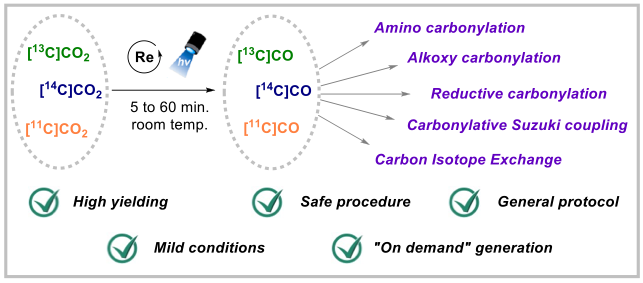
-to-CO conversion remains an often-overlooked criterion that prevents a productive and direct valorisation of CO into high-value-added chemicals. Herein, we report a photocatalytic process that unlocks full and fast CO2-to-CO conversion (< 10 minutes) and its straightforward valorisation into human health related field of radiochemistry with carbon isotopes. Guided by reaction-model-based kinetic simulations to rationalize reaction optimisations, this manifold opens new opportunities for the direct access to 11C- and 14C-labeled pharmaceuticals from their primary isotopic sources [11C]CO2 and [14C]CO2.
Efficient Hydrogen Production at pH 7 in Water with a Heterogeneous Electrocatalyst Based on a Neutral Dimeric Cobalt-Dithiolene Complex. C. Zhang, E. Prignot, O. Jeannin, A. Vacher, D. Dragoe, F. Camerel,* Z. Halime,* R. Gramage-Doria* (2023) ACS Catalysis 13, 2367-2373
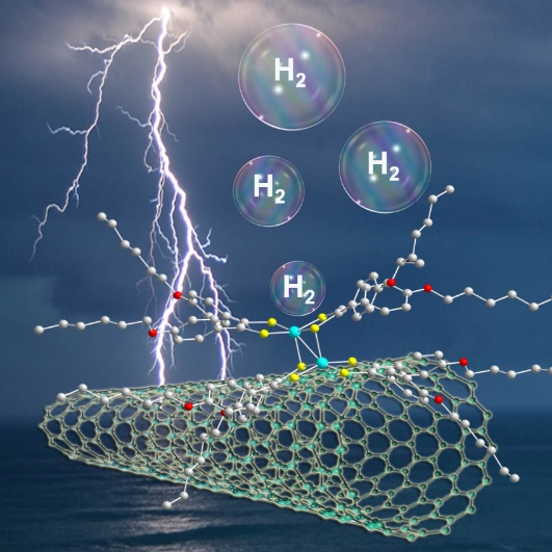
The development of efficient hydrogen production technologies is fundamental for global sustainability. As such, electrocatalysts derived from earth abundant metal complexes are appealing, and interesting performances have typically been disclosed under acidic conditions in organic solvents. However, their applicability under relevant pH neutral conditions is underexplored. Herein, we demonstrate that non-ionic, dimeric cobalt-dithiolene complexes supported on multi-wall carbon nanotubes (MWCNTs)/carbon paper (CP) electrode are powerful electrocatalysts for hydrogen production in aqueous media at pH 7. The high turnover numbers encountered (TON up to 50980) after long reaction times (up to 16 hours) are reasoned by the increased electro-active cobalt concentration on the modified electrode, which is ca. 4 times higher than that of a state-of-the-art cobalt porphyrin electrocatalyst. These findings point out that immobilizing well-defined, multinuclear low-cost metal complexes on carbon material is a promising strategy to design highly electroactive electrodes.
Bio-Inspired Bimetallic Cooperativity Through a Hydrogen Bonding Spacer in CO2 Reduction. C. Zhang, P. Gotico, R. Guillot, D. Dragoe, W. Leibl, Z. Halime,* A. Aukauloo* (2023) Angew. Chem. Int. Ed. 62, e202214665
At the core of carbon monoxide dehydrogenase (CODH) active site two metal ions together with hydrogen bonding scheme from amino acids orchestrate the interconversion between CO2 and CO. We have designed a molecular catalyst implementing a bimetallic iron complex with an embarked second coordination sphere with multi-point hydrogen-bonding interactions. We
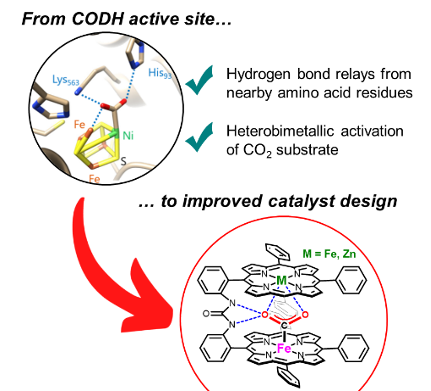

found that, when immobilized on carbon paper electrode, the dinuclear catalyst enhances up to four-fold the heterogeneous CO2 reduction to CO in water with an improved selectivity and stability compared to the mononuclear analogue. Interestingly, quasi-identical catalytic performances are obtained when one of the two iron centers was replaced by a redox inactive Zn metal, questioning the cooperative action of the two metals. Snapshots of X-ray structures indicate that the two metalloporphyrin units tethered by a urea group is a good compromise between rigidity and flexibility to accommodate CO2 capture, activation, and reduction.
Dissection of Light-Induced Charge Accumulation at a Highly Active Iron Porphyrin: Insights in the Photocatalytic CO2 Reduction. E. Pugliese, P. Gotico, I. Wehrung, B. Boitrel, A. Quaranta, M.H. Ha-Thi, T. Pino, M. Sircoglou, W. Leibl, Z. Halime,* A. Aukauloo* (2022) Angew. Chem. Int. Ed. 61, e202117530

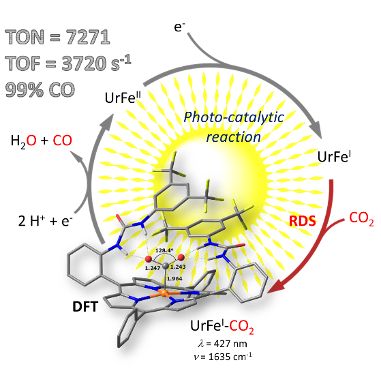
Using light energy to drive multielectron catalytic reduction of CO2 to produce a fuel is a contemporary research thrust. Recent efforts in the design of porphyrin-based catalysts have positioned them among the most performant electrocatalysts for CO2 reduction. Powering these catalysts with the help of photosensitizers to realize light-driven catalysis comes along with a couple of
unsolved challenges that need to be addressed with much vigor. We have designed an iron porphyrin catalyst decorated with urea functions (UrFe) acting as a multipoint hydrogen bonding scaffold towards the CO2 substrate. When powered by a molecular photosensitizer in presence of an electron donor and water, a spectacular photocatalytic activity reaching unreported TONs and TOFs as high as 7270 and 3720 h-1 respectively are observed. Importantly, we have spectroscopically characterized all the photo-reduced FeII, FeI and Fe0 states and compared with the electrochemically generated ones. While the Fe0 redox state has been widely accepted as the catalytically active species, we show here that the FeI species is already involved in the CO2 activation which represents the rate determining step in the photocatalytic cycle. DFT calculations bring support to our experimental findings that constitute ahis new paradigm in the catalytic reduction of CO2.
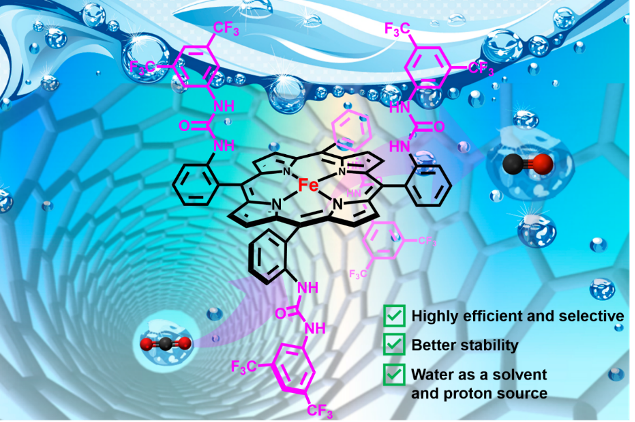
Second-sphere hydrogen-bonding enhances heterogeneous electrocatalytic CO2 to CO reduction by iron porphyrins in water. C. Zhang, D. Dragoe, F. Brisset, B. Boitrel, B. Lassalle-Kaiser, W. Leibl, Z. Halime,* A. Aukauloo* (2021) Green Chemistry 23, 8979-8987
Intense efforts are currently being injected in discovering cost-effective catalysts for the selective reduction of carbon dioxide (CO2). Much advances have indeed been realized in the design of molecular complexes containing second coordination chemical functionalities that have contributed to boost the homogeneous electrocatalytic activity. Implementing these chemical facets in heterogeneous catalysis can give access to an improved catalysis while opening at the same time the way for practical and sustainable applications. Here we report the catalytic properties for CO2 electroreduction of a
Through-Space Electrostatic Interactions Surpass Classical Through-Bond Electronic Effects in Enhancing CO2 Reduction Performance of Iron Porphyrins. A. Khadhraoui, P. Gotico, W. Leibl, Z. Halime,* A. Aukauloo,* (2021) ChemSusChem 14, 1308
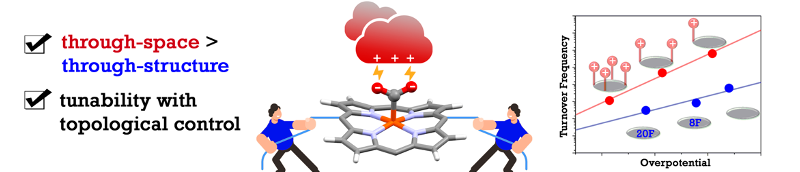
In his pioneering work to unravel the catalytic power of enzymes, Warshel has pertinently validated that electrostatic interactions play a major role in the activation of substrates. Implementing such chemical artifice in molecular catalysts may help improve their catalytic properties. In this study, we have designed a series of tetra-, di- and mono-substituted iron porphyrins with cationic imidazolium groups. Their presence in the second coordination sphere help stabilize the [Fe-CO2] intermediate through electrostatic interactions. We found herein that the electrocatalytic overpotential is a function of the number of embarked imidazolium. Importantly, we evidenced a gain of six orders of magnitude in turnover frequencies going from a tetra- to a mono-substituted catalyst. Furthermore, our comparative study shows that catalytic performances trend of through-space electrostatic interaction, a first topological effect reported for iron porphyrins, outperforms the classical through-structure electronic effect.

Atropisomeric Hydrogen Bonding Control for CO2 Binding and Electrocatalytic Reduction Enhancement at Iron Porphyrins. P. Gotico, L. Roupnel, R. Guillot, M. Sircoglou, W. Leibl, Z. Halime,* A. Aukauloo,* (2020) Angew. Chem. Inter. Ed. 59, 22451
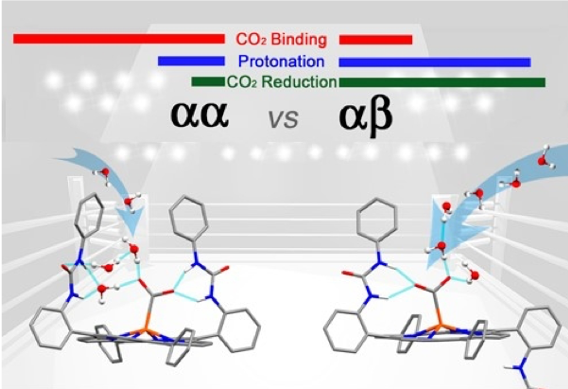
The manipulation of the second coordination sphere for improving the electrocatalytic CO2 reduction has led to amazing breakthroughs with hydrogen bonding, local proton source, or electrostatic effects. We have developed two atropisomers of an iron porphyrin complex holding two urea functions acting as multiple hydrogen bonding tweezers to lock the metal bound CO2 in a similar fashion found in the carbon monoxide dehydrogenase (CODH) enzyme. We found that the aa topological isomer with the two urea groups on the same side of the porphyrin platform provides a stronger binding affinity to tether
the incoming CO2 substrate in comparison to the ab disposition. However, the electrocatalytic activity of the ab atropisomer outperforms its congener with one of the highest reported turnover frequency at low overpotential. The strong H/D KIE observed for the aa system indicates the existence of a tight water hydrogen bonding network for proton delivery which is disrupted upon addition of exogenous acid source. While the small H/D KIE for the ab isomer and the enhanced electrocatalytic performance upon addition of stronger acid pertain the free access of protons to the bound CO2 on the opposite side of the urea arm.
Recent advances in metalloporphyrin-based catalyst design towards carbon dioxide reduction: from bio-inspired second coordination sphere modifications to hierarchical architectures. P. Gotico, Z. Halime,* A. Aukauloo,* (2020) Dalton Trans. 49, 2381

Research in the development of new molecular catalysts for the selective transformation of CO2 to reduced forms of carbon is attracting enormous interest from chemists. Molecular catalyst design hinges on the elaboration of ligand scaffolds to manipulate the electronic and structural properties for the fine tuning of the reactivity pattern. A cornucopia of ligand sets has been designed along this line and more and more are being reported. In this quest, the
porphyrin molecular platform has been under intensive focus due to the unmatched catalytic properties of metalloporphyrins. There have been rapid advances in this particular field during the last few years wherein both electronic and structural aspects in the second coordination spheres have been addressed to shift the overpotential and improve the catalytic rates and product selectivity. Metalloporphyrins have also attracted much attention in terms of the elaboration of hybrid materials for heterogeneous catalysis. Here too, some promising activities have made metalloporphyrin derivatives serious candidates for technological implementation. This review collects the recent advances centered around the chemistry of metalloporphyrins for the reduction of CO2.
Spectroscopic Characterization of Bio-inspired Ni-based Proton Reduction Catalyst Bearing Pentadentate N2S3 Ligand with Improved Photocatalytic Activity. P. Gotico, D. Moonshiram, C. Liu, X. Zhang, R. Guillot, A. Quaranta, Z. Halime, W. Leibl, A. Aukauloo (2020) Chem. Eur. J. 26, 2859-2868
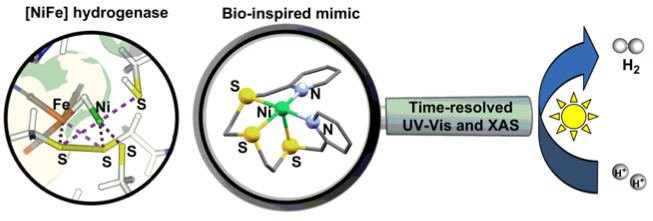
Inspired by the sulphur-rich environment of [NiFe] hydrogenase, a novel Ni-based proton reduction catalyst bearing an acyclic pentadentate N2S3 ligand has been shown to improve photocatalytic activity and has been mechanistically probed by time-resolved optical and X-ray absorption spectroscopy.

Second‐Sphere Biomimetic Multipoint Hydrogen‐Bonding Patterns to Boost CO2 Reduction of Iron Porphyrins. P. Gotico, B. Boitrel, R. Guillot, M. Sircoglou, A. Quaranta, Z. Halime,* W. Leibl, A. Aukauloo,* (2019) Angew. Chem. Inter. Ed. 58, 4504-4509 (10.1002/anie.201814339)
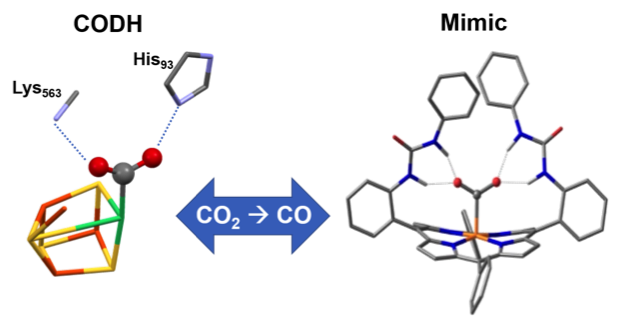
Like CODH: Urea groups at an iron porphyrin catalyst give multipoint hydrogen‐bonding stabilization resembling that found in the CO2 adduct of carbon monoxide dehydrogenase (CODH). The catalyst gave improved CO2 reduction to CO. Entrapped water molecules within the molecular clefts were found to be the source of protons for the CO2 reduction.

Local Ionic Liquid Environment at a Modified Iron Porphyrin Catalyst Enhances Electrocatalytic Performance of CO2 to CO Reduction in Water. A. Khadhraoui, P. Gotico, B. Boitrel, W. Leibl, Z. Halime,* A. Aukauloo,* (2018) Chem. Comm. 54, 11630-11633
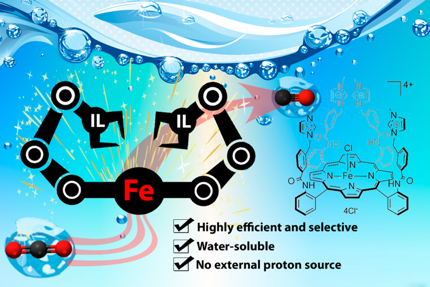
In this study, we report a strategy to attach methylimidazolium fragments as ionic liquid units on an established iron porphyrin catalyst for the selective reduction of CO2 to CO. Importantly, we found that the tetra-methylimidazolium containing porphyrin exhibits an exalted electrocatalytic activity at low overpotential in water precluding the need for an external proton donor.
Visible-Light-Driven Reduction of CO2 to CO and Its Subsequent Valorization in Carbonylation Chemistry and 13C Isotope Labeling. P. Gotico, A. Del Vecchio, D. Audisio, A. Quaranta, Z. Halime,* W. Leibl, A. Aukauloo,* (2018) ChemPhotoChem 2, 715-719
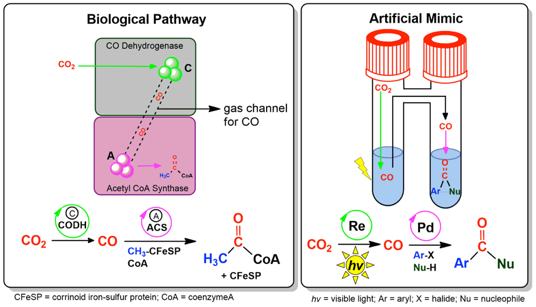
Reduce, reuse, recycle: A CO2 valorization strategy based on its photocatalytic reduction to CO and the direct use of the produced CO in a palladium‐catalyzed aminocarbonylation reaction is reported. This approach provides a means for more efficient application of 13C‐isotope and 14C‐radioisotope‐labeled CO2 in pharmaceutically relevant drug labeling.

For more details, see the comprehensive list of our publications HERE.
Last update on August 1st 2023