Artificial photosynthesis
Axis 3 - OXIDATION REACTIONS
Led by Marie Sircoglou
Photocatalytic generation of a non-heme Fe(III)-hydroperoxo species with O2 in water for the oxygen atom transfer reaction. E. Pugliese, N.T. Vo, A. Boussac, F. Banse, Y. Mekmouche, J. Simaan, T. Tron, P. Gotico, M. Sircoglou, Z. Halime, W. Leibl, A. Aukauloo (2022) Chemical Science 13, 12332-12339
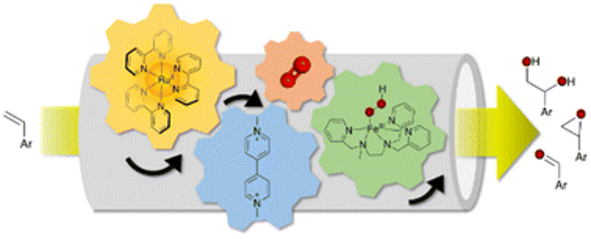
Coupling a photoredox module and a bio-inspired non-heme model to activate O2 for oxygen atom transfer (OAT) reaction requires a vigorous investigation to shed light on the multiple competing electron transfer steps, charge accumulation and annihilation processes, and the activation of O2 at the
catalytic unit. We found that the efficient oxidative quenching mechanism between [Ru(bpy)3]2+ chromophore and a reversible electron mediator, methyl viologen (MV2+), to form the reducing species methyl viologen radical (MV•+) can convey an electron to O2 to form the superoxide radical and resetting an Fe(III) species in a catalytic cycle to the Fe(II) state in an aqueous solution. The formation of the Fe(III)-hydroperoxo (FeIII- OOH) intermediate therefrom to evolve to highly oxidized iron-oxo species to perform the OAT reaction to an alkene substrate. Such a strategy allows to bypass the challenging task of charge accumulation at the molecular catalytic unit for the two-electron activation of O2. The FeIII-OOH catalytic precursor was trapped and characterized by EPR spectroscopy pertaining a metal assisted catalysis. Importantly, we found that the substrate itself can act as the electron donor to reset the photooxidzed chromophore in the initial state closing the photocatalytic loop hence excluding the use of a sacrificial electron donor. Laser Flash Photolysis (LFP) studies and spectroscopic monitoring during photocatalysis lend credence to the proposed catalytic cycle.
Ligand Radical Mediated Water Oxidation by a Family of Copper o-Phenylene Bis-oxamidate Complexes. S. Chattopadhyay, A. Ghatak, Y. Ro, R. Guillot, Z. Halime, A. Aukauloo, A. Dey (2021) Inorg. Chem. 60, 9442-9445
Understanding the reactivity landscape for the activation of water till the formation of the O-O bond and O2 release in molecular chemistry is a decisive step in guiding the elaboration of cost-effective catalysts for the oxygen-evolving reaction (OER). Copper (II) complexes have recently caught the attention of chemists as catalysts for the 4e-/4H+ water oxidation process. While a copper (IV) intermediate has been proposed as the reactive intermediate species, yet no spectroscopic signature has been reported so far. Copper (III) ligand radical species have also been formulated and supported by theoretical studies. We found, herein, that the reactivity sequence for the water oxidation with a family of Copper (II) o-phenylene bisoxamidate complexes are a function of the substitution pattern on the periphery of the
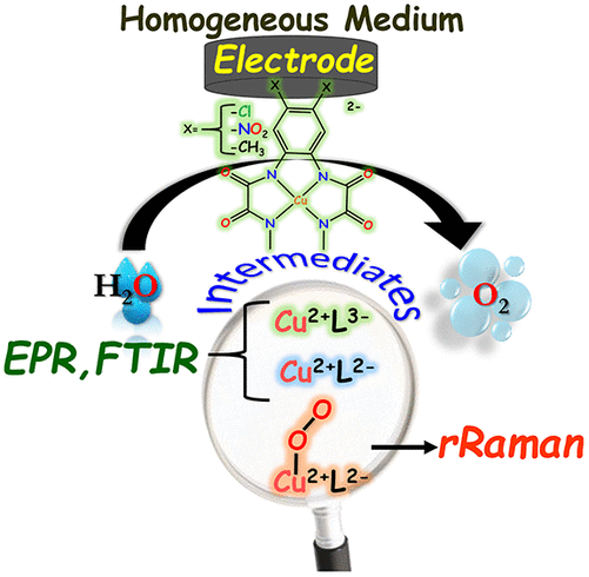
aromatic ring. In-situ IR, EPR and rR spectroelectrochemical studies helped to sequence the elementary electrochemical and chemical events leading towards the O2 formation selectively at the copper center. A copper (II) superoxide species is identified as the reactive intermediate, the stability of which governs the selectivity of 4e- oxidation of water to molecular oxygen.
Rhodium(II)-catalyzed enantioselective intermolecular aziridination of alkenes. V. Boquet, A. Nasrallah, A. L. Dana, E. Brunard, P. H. Di Chenna, F. J. Duran, P. Retailleau, B. Darses, M. Sircoglou, P. Dauban (2022) J. Am. Chem. Soc. 144, 17156–17164
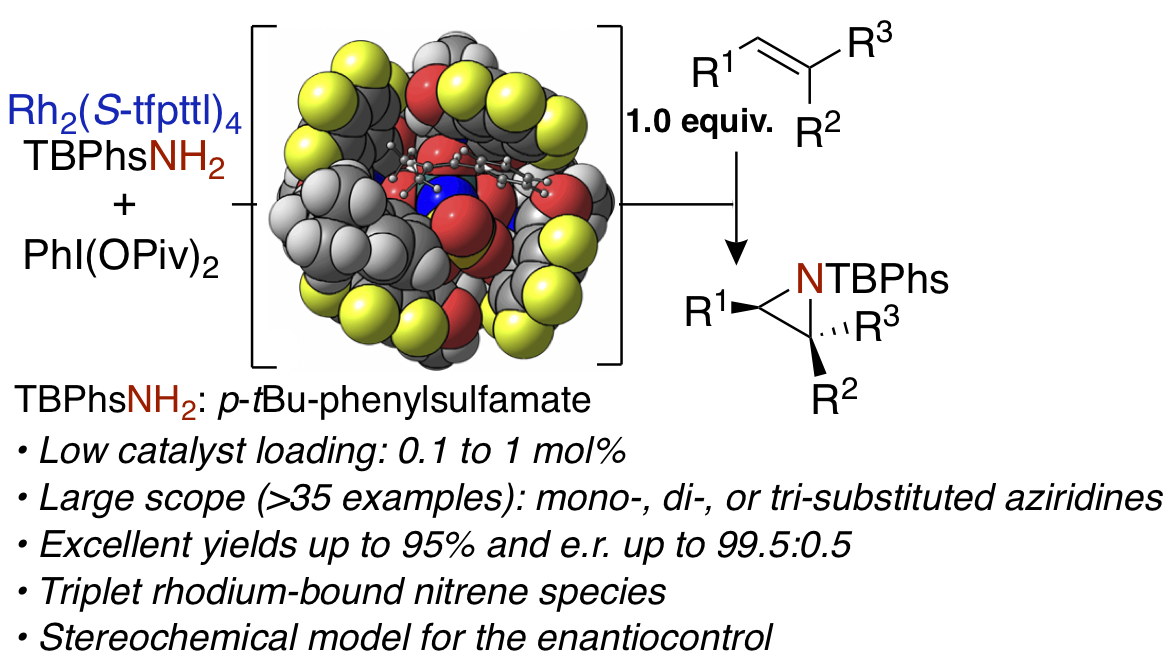
C4-Symmetrical dirhodium(II) tetracarboxylates are highly efficient catalysts for the asymmetric intermolecular aziridination of substituted alkenes with sulfamates. The reaction proceeds with high levels of efficiency and chemoselectivity to afford aziridines with excellent yields of up to 95% and enantiomeric excesses of up to 99%. The scope of the alkene aziridination includes mono-, di-, and trisubstituted olefins as well as the late-stage functionalization of complex substrates. The reaction
can be performed on a gram-scale with a catalyst loading of 0.1 mol %. Our DFT study led us to propose a two-spin-state mechanism, involving a triplet Rh−nitrene species as key intermediate to drive the stereocontrolled approach and activation of the substrate.
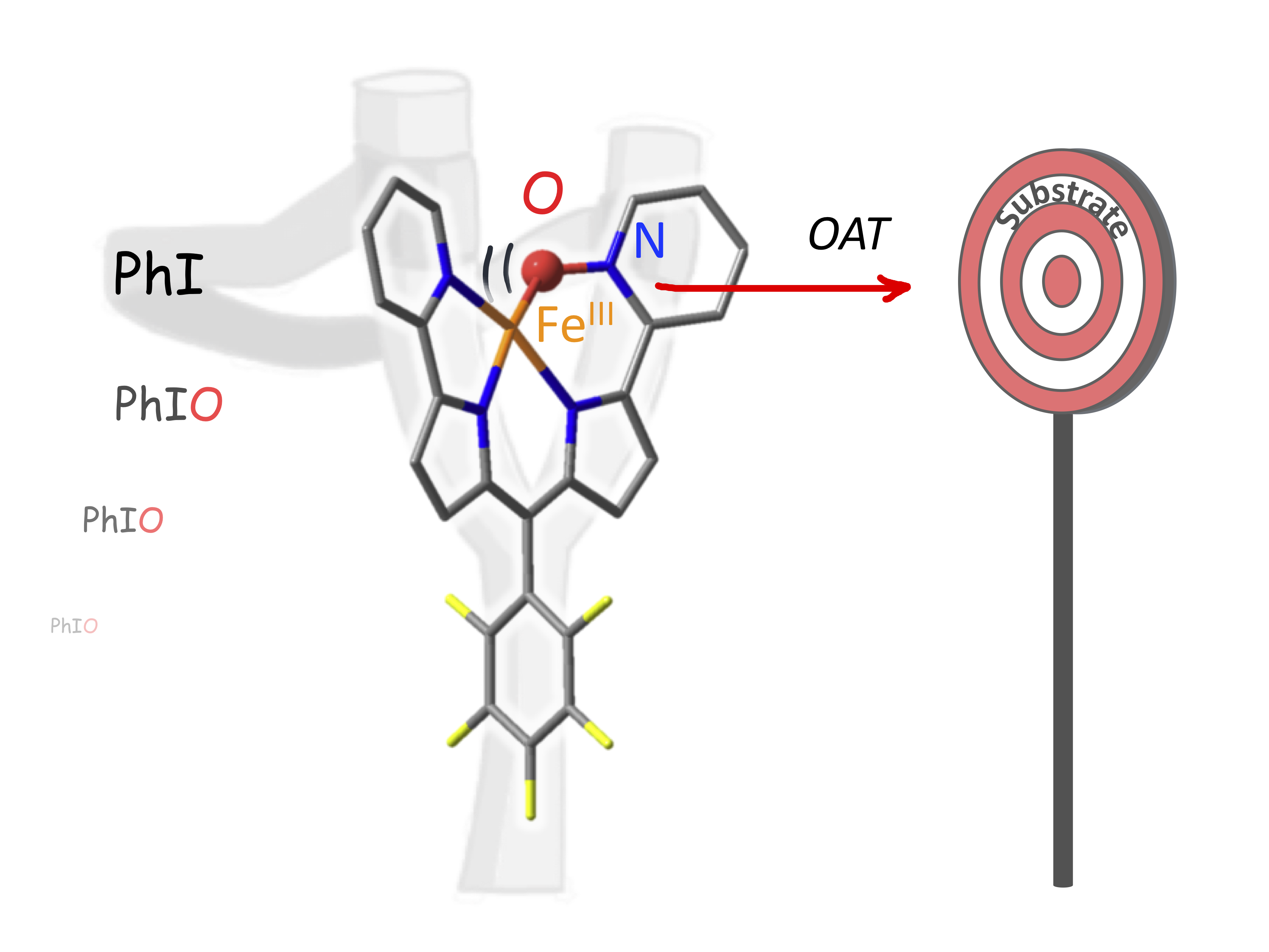
Intercepting a transient non-hemic pyridine N-oxide Fe(III) species involved in OAT reactions. T. Vo, C. Herrero, R. Guillot, T. Inceoglu, W. Leibl, M. Clémancey, P. Dubourdeaux, G. Blondin, A. Aukauloo, M. Sircoglou (2021) Chem. Comm. 57, 12836-12839
In the context of bioinspired OAT catalysis, we developed a tetra- dentate dipyrrinpyridine ligand, a hybrid of hemic and non-hemic models. The catalytic activity of the iron(III) derivative was investigated in the presence of iodosylbenzene. Unexpectedly, MS, EPR, Mo ̈ssbauer, UV-visible and FTIR spectroscopic signatures supported by DFT calculations provide convincing evidence for the involvement of a relevant FeIII–O–NPy active intermediate.
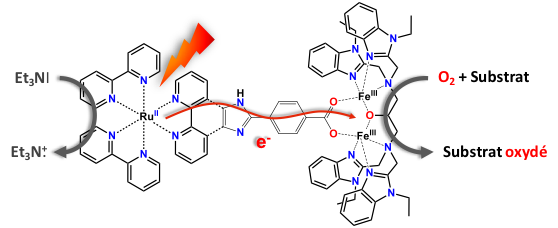
A Reversible Electron Relay to Exclude Sacrificial Electron Donors in the Photocatalytic Oxygen Atom Transfer Reaction with O2 in Water. N. T. Vo, Y. Mekmouche, T. Tron, R. Guillot, F. Banse, Z. Halime, M. Sircoglou, W. Leibl, A. Aukauloo (2019) Angew. Chem. Inter. Ed. 58,16023-16027
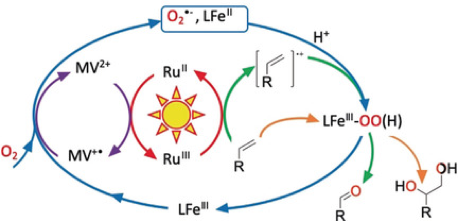
The evil necessity of sacrificial electron donors (SEDs) in the photoactivation of O2 can be ousted by the use of a reversible electron acceptor (methylviologen). It intervenes as an electron carrier from the excited photosensitizer to reduce both O2 and an iron(III) catalyst to form a reactive FeIII–peroxo species for oxygen atom transfer reactions. Oxidation of an organic substrate closes the photocatalytic cycle.
Light-Induced Activation of the Du Bois [RhII2(Esp)2] Catalyst for Nitrogen Atom Transfer Reactions. R. Farran, C. Ducloiset, J. Buendia, N. T. Vo, R. Guillot, Z. Halime, P. Dauban, W. Leibl, M. Sircoglou, A. Aukauloo (2017) ChemPhotoChem 1, 562-567
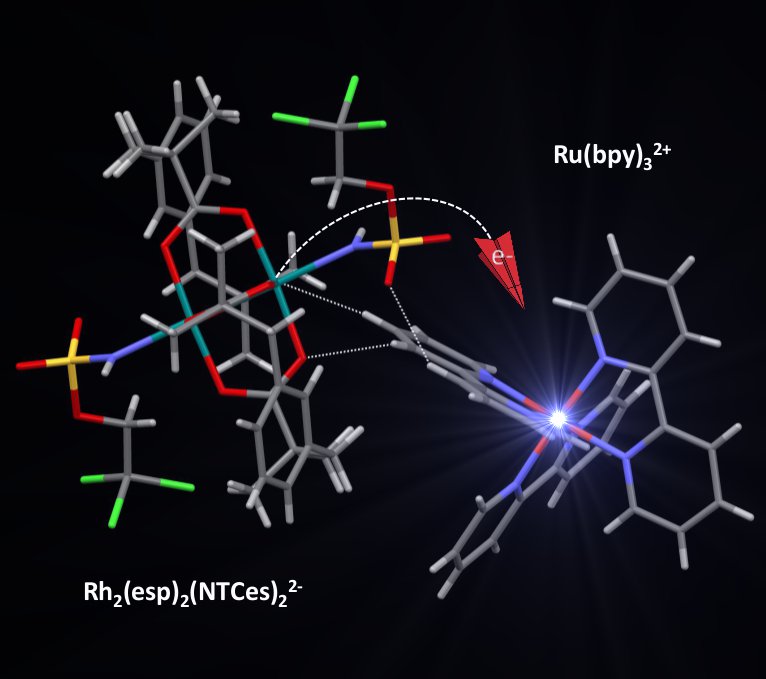
[Rh2(Esp)2] is a well-established catalyst for intra- and intermolecular C(sp)3H bond amination. We have prepared and isolated complex I, [Rh2(Esp)2(NHTces)2]2, where the catalyst bears sul- fonamidate ligands (NHTces = NHSO3CH2CCl3) as nitrogen sources in the coordination sphere of the rhodium centre. The possibility for light activation instead of chemical activation of the [Rh2(Esp)2] catalyst and complex I is demonstrated using [Ru(bpy)3]2+ as the photosensitizer. The oxidation of the two dinuclear Rh complexes occurs through two distinct pathways. On one hand, [Rh2(Esp)2] is oxidized in a bimolecular reaction with the RuIII species formed after excitation of the photosensi- tizer in the presence of an electron
acceptor. In contrast, the lower oxidation potential of complex I allows for its oxidation to occur directly from the excited state of the photosensitizer, helped by ion-pair formation between the negatively charged complex I with [Ru(bpy)3]2+. The charge-separated state of the oxidized complex I / reduced photosensitizer had a lifetime of 50 μs.
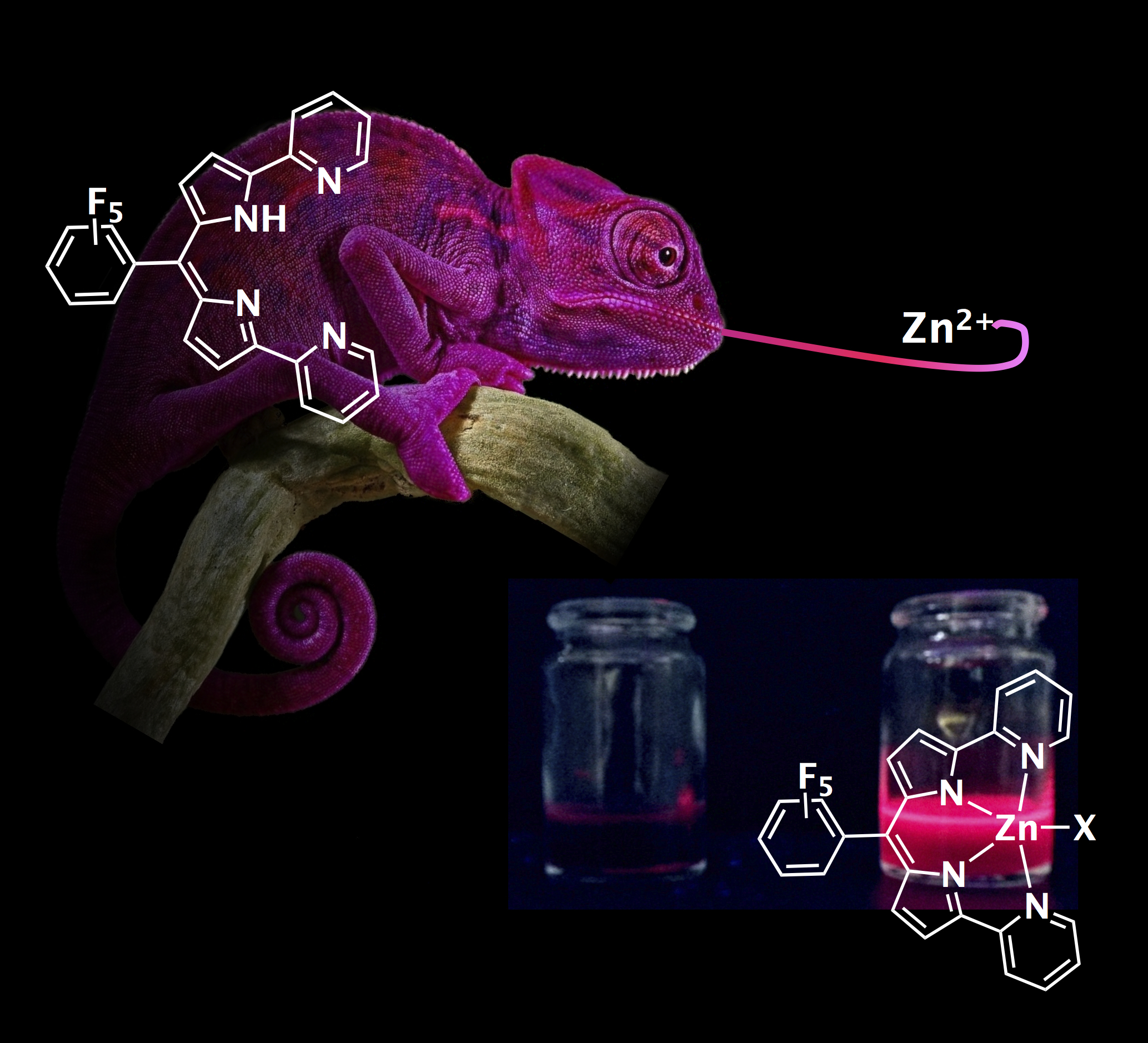
Monoanionic Dipyrrin–Pyridine Ligands: Synthesis, Structure and Photophysical Properties. C. Ducloiset, P. Jouin, E. Paredes, R. Guillot, M. Sircoglou, M. Orio, W. Leibl, A. Aukauloo (2015)
A novel monoanionic tetradentate N4 ligand (F5DPPy) based on a dipyrromethene skeleton as a molecular platform and decorated with pyridine rings at the 1- and 9-positions of the dipyrrin motif has been prepared and characterized. Interestingly, although this ligand is weakly fluorescent, it presents a chelation-enhanced fluorescence effect of around 150 times upon coordination to ZnII. Time-dependent (TD) DFT calculations reproduce nicely the spectroscopic features of both the ligand and the complex, and analysis of the electron den- sity redistribution in the excited state suggests that a better orbital overlap of the HOMO and LUMO in F5DPPyZnCl compared with F5DPPy is responsible for the more intense transitions observed with the former system. As such, this ligand opens interesting perspectives in the design of ratiometric sensors.
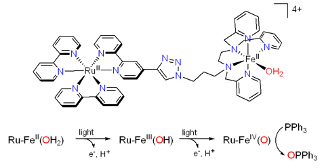
Successive light-induced two electron transfers in a Ru-Fe supramolecular assembly: From Ru-Fe(II)-OH2 to Ru-Fe(IV)-oxo. C. Herrero, A. Quaranta, M. Sircoglou, K. Sénéchal-David, A. Baron, I. M. Marín, C. Buron, J.-P. Baltaze, W. Leibl, A. Aukauloo, F. Banse Chem. Science 6, 2323-2327
In the present work we describe the synthesis and study of a RuII–FeII chromophore–catalyst assembly designed to perform the light-induced activation of an iron bound water molecule and subsequent photo-driven oxidation of a substrate. Using a series of spectroscopic techniques, we demonstrate that excitation of the chromophore unit with 450 nm light, in the presence of a sacrificial electron acceptor, triggers a cascade of electron transfers leading to the formation of a high valent iron(IV)–oxo center from an iron(II)-bound water molecule. The activity of this catalytic center is illustrated by the oxidation of triphenyl phosphine.
Photoassisted Generation of a Dinuclear Iron(III) Peroxo Species and Oxygen-Atom Transfer. F. Avenier, C. Herrero, W. Leibl, A. Desbois, R. Guillot, J-P. Mahy, A. Aukauloo (2013) Angew. Chem. Int. Ed. 52, 3634-3637
Oxidation processes usually necessitate powerful oxidizing reagents such as hydrogen peroxide. As an alternative, we reported the photocatalytic activation of O2 at a diiron(II) complex - ruthenium(II) photosensitizer dyad where the dinuclear iron complex mimics the methane monooxygenase catalytic center. This biomimetic approach allowed to photooxidize organic substrates in the absence of strong oxidant by using dioxygen as the sole source of O atom.
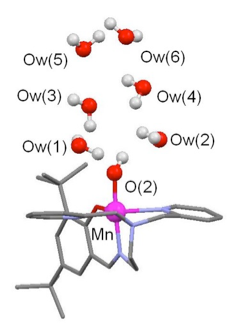
Implications of remote water molecules on the electron transfer coupled processes at a nonporphyrinic Mn(III)-hydroxido complex. S. El Ghachtouli, B. Lassalle-Kaiser, P. Dorlet, R. Guillot, E. Anxolabéhère-Mallart, C. Costentin, A. Aukauloo (2011) Energy Environ. Sci. 4, 2041-2044
Despite recent advances in the field of molecular catalysis, noble metals based complexes are still the best candidates to promote water oxidation. We are dedicated to understand the factors that govern the activation of a water molecule at more earth abundant metal centers as in nature. We have shown, for instance, the implication of surrounding water molecules in the formation of highly oxidized Mn-oxo species. As part of the we also managed to photoinduce the activation of a water molecule in a photosensitizer-catalyst dyad to photooxydize a substrate using water as the sole source of O.
For more details, see the comprehensive list of our publications HERE.
Last update on August 1st 2023