Photosynthèse artificielle
Axe 1 - TRANSFERTS D'ELECTRONS PHOTOINDUITS
Animé par Ally Aukauloo (ally.aukauloo@universite-paris-saclay.fr)
Notre biosphère est alimentée en énergie par la lumière provenant du soleil. La nature a développé des antennes très efficaces pour capter et transformer cette énergie en potentiel chimique. Ce dernier est ensuite utilisé pour catalyser l’oxydation de l’eau d’une part et la réduction du CO2 d’autre part. Mettre au point des systèmes synthétiques capables de reproduire les premiers événements de transfert d’électron photoinduit et les coupler à la catalyse est primordial pour le développement de la photosynthèse artificielle.
Proton-controlled Action of an Imidazole as Electron Relay in a Photoredox Triad. P. Gotico, C. Herrero, S. Protti, A. Quaranta, S. Sheth, R. Fallahpour, R. Farran, Z. Halime, M. Sircoglou, A. Aukauloo, W. Leibl (2022) Photochemical and photobiological Sciences 21, 247
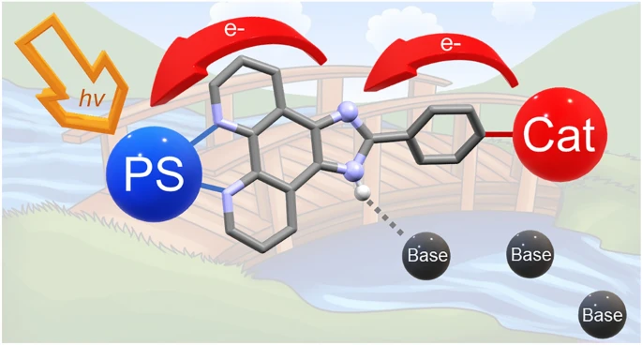
Electron relays play a crucial role for efficient light-induced activation by a photo-redox moiety of catalysts for multi-electronic transformations. Their insertion between the two units reduces detrimental energy transfer quenching while establishing at the same time unidirectional electron flow. This rectifying function allows charge accumulation necessary for catalysis. Mapping these events in photophysical studies is an
important step towards the development of efficient molecular photocatalysts. Three modular complexes comprised of a Ru-chromophore, an imidazole electron relay function, and a terpyridine unit as coordination site for a metal ion were synthesized and the light-induced electron transfer events studied by laser flash photolysis. In all cases, formation of an imidazole radical by internal electron transfer to the oxidized chromophore was observed. The effect of added base evidenced that the reaction sequence depends strongly on the possibility for deprotonation of the imidazole function in a proton-coupled electron transfer process. In the complex with MnII present as a proxy for a catalytic site, a strongly accelerated decay of the imidazole radical together with a decreased rate of back electron transfer from the external electron acceptor to the oxidized complex was observed. This transient formation of an imidazolyl radical is clear evidence for the function of the imidazole group as an electron relay. The implication of the imidazole proton and the external base for the kinetics and energetics of the electron trafficking is discussed.
Phthalocyanine as a Bioinspired Model for Chlorophyll f-Containing Photosystem II Drives Photosynthesis into the Far-Red Region. J. Follana-Berná, R. Farran, W. Leibl, A. Quaranta, Á. Sastre-Santos, A. Aukauloo (2022) Angewandte Chemie International Edition 60, 12284
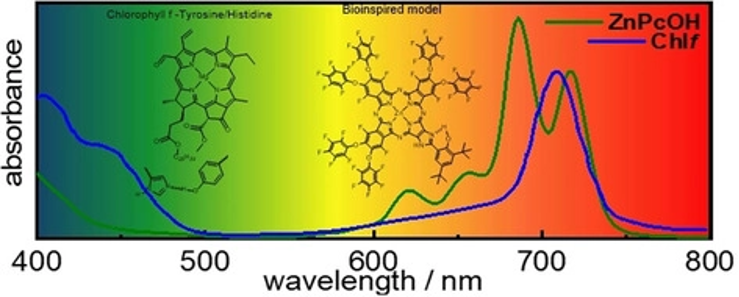
The textbook explanation that P680 pigments are the red limit to drive oxygenic photosynthesis must be reconsidered by the recent discovery that chlorophyll f (Chlf)-containing Photosystem II (PSII) absorbing at 727 nm can drive water oxidation. Two different families of unsymmetrically substituted Zn phthalocyanines (Pc) absorbing in the 700–800 nm spectral window and containing a fused imidazole-phenyl substituent or a fused imidazole-hydroxyphenyl group have been synthetized and

characterized as a bioinspired model of the Chlf/TyrosineZ/Histidine190 cofactors of PSII. Transient absorption studies in the presence of an electron acceptor and irradiating in the far-red region evidenced an intramolecular electron transfer process. Visible and FT-IR signatures indicate the formation of a hydrogen-bonded phenoxyl radical in ZnPc II-OH. This study sets the foundation for the utilization of a broader spectral window for multi-electronic catalytic processes with one of the most robust and efficient dyes.
Enhanced Photoinduced Electron Transfer Through a Tyrosine Relay in a De Novo Designed Protein Scaffold Bearing a Photoredox Unit and a FeIIS4 Site. A. Tebo, A. Quaranta, V. L. Pecoraro, A. Aukauloo (2021) ChemPhotoChem 5 : 665-668
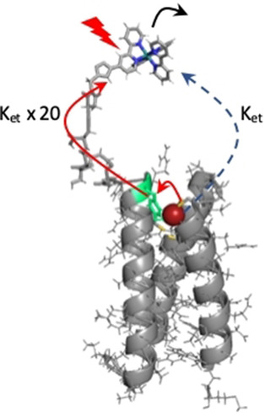
We have designed an electron transfer chain incorporated into a de novo protein scaffold, which is capable of photoinduced intramolecular electron transfer between a photoredox unit and a FeIIS4 site through a tyrosine amino acid relay. The kinetics were characterized by nanosecond laser pulse photolysis and revealed that electron transfer from the photoredox unit [RuIIIbpymal]3+ proceeds most efficiently via a tyrosine located circa 16 Å from Rubpymal ((bpymal=1-((1-([2,2′-bipyridin]-4-yl)-1H-1,2,3-triazol-4-yl)methyl)-1H-pyrrole-2,5-dione)). Removal of the tyrosine as the electron relay station results in a 20-fold decrease in the apparent rate constant for the electron transfer.
Tracking light-induced electron transfer toward O2 in a hybrid photoredox-laccase system. R. Farran, Y. Mekmouche, N. T. Vo, C. Herrero, A. Quaranta, M. Sircoglou, F. Banse, P. Rousselot-Pailley, A. J. Simaan, A. Aukauloo, T. Tron, W. Leibl (2021) iScience 24, 102378
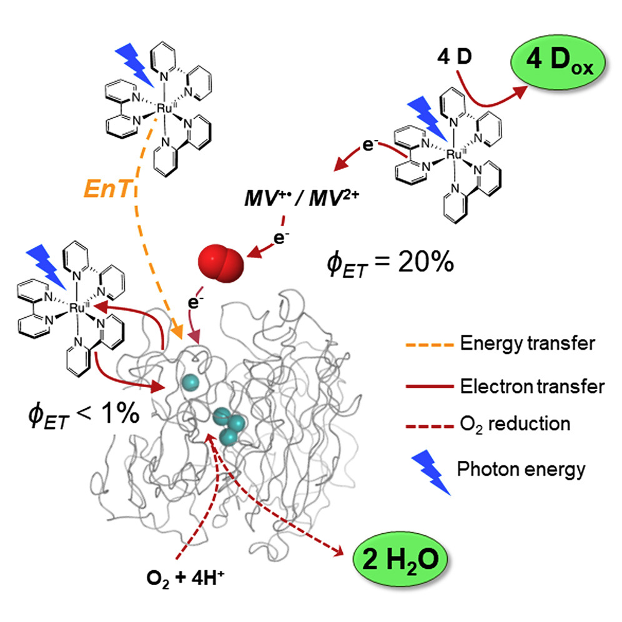
Photobiocatalysis uses light to perform specific chemical transformations in a selective and efficient way. The intention is to couple a photoredox cycle with an enzyme performing multielectronic catalytic activities. Laccase, a robust multicopper oxidase, can be envisioned to use dioxygen as a clean electron sink when coupled to an oxidation photocatalyst. Here, we provide a detailed study of the coupling of a [Ru(bpy)3]2+ photosensitizer to laccase. We demonstrate that efficient laccase reduction requires an electron relay like methyl viologen. In the presence of dioxygen, electrons transiently stored in superoxide ions are scavenged by laccase to form water instead of H2O2. The net result is the photo accumulation of highly oxidizing [Ru(bpy)3]3+. This study provides ground for the use of laccase in tandem with a light-driven oxidative process and O2 as one-electron transfer relay and as four-electron substrate to be a sustainable final electron acceptor in a photocatalytic process.
Modèle de la TyrZ-His190
Le Photosystème II (PSII) est l’enzyme responsable de la photooxydation de l’eau en dioxygène. Si les chemins de transport d’électron à la fois à l’intérieur et à l’extérieur du PSII sont plutôt bien élucidés, le transport des protons reste encore énigmatique. Nous avons développé un modèle bioinspiré de la paire d’acides aminés tyrosine Z - histidine 190, qui est connue pour son rôle de relais électronique entre le pigment chlorophyllien et le centre catalytique du dégagement d’oxygène (CDO). Les résultats, publiés dans la revue Angewandte Chemie International Edition, révèlent que cette paire d’acides aminés jouerait aussi un rôle crucial dans l’évacuation des protons du CDO. Cette découverte vient conforter les récentes propositions mécanistiques qui s’appuient sur une structure cristallographique à très haute résolution du PSII. Celles-ci impliquent l’existence d’un canal à proton formé d’un réseau de molécules d’eau interagissant avec le CDO et ces deux acides aminés.
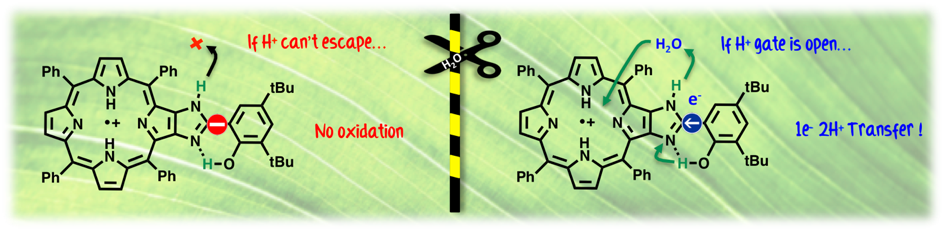
Ce travail est le fruit d’une collaboration avec les chercheurs de l’Institut de Biologie Intégrative de la Cellule (CNRS/Université Paris-Sud/CEA), de l’Institut des Sciences Moléculaires de Marseille (CNRS/Aix-Marseille Université/Centrale Marseille), du synchrotron SOLEIL (CNRS/CEA) ainsi que l’Université d’Héraklion Crète.
Water Molecules Gating a Photoinduced One Electron Two Protons Transfer in a Tyr/His model of Photosystem II. G. Chararalambidis, S. Das, A. Trapali, A. Quaranta, M. Orio, Z. Halime, P. Fertey, R. Guillot, C. Athanassios, W. Leibl, A. Aukauloo, M. Sircoglou (2018) Angew. Chem. Int. Ed. 57, 9013-9017
Accumulation de charge à 2 électrons
En collaboration avec des équipes de l’Institut des Sciences Moléculaires d’Orsay (CNRS/Université Paris-Sud, Université Paris-Saclay), et de l’Institut de Biologie Intégrative de la Cellule (CNRS/CEA Paris-Saclay, Université Paris-Saclay) nous avons franchi une étape supplémentaire dans le processus de photoaccumulation à 2 électrons. Nous avons en effet réussi à suivre les événements induits par l’absorption d’un deuxième photon permettant le transfert d’un deuxième électron par spectroscopie. La détection, directe et sans équivoque d’un état à double transfert de charge stockant une énergie de 1.1 eV, est une première dans une dyade moléculaire. Nous avons de plus mesuré une durée de vie de 0.2 ms pour cet état, valeur exceptionnellement longue pour un système moléculaire. Ces
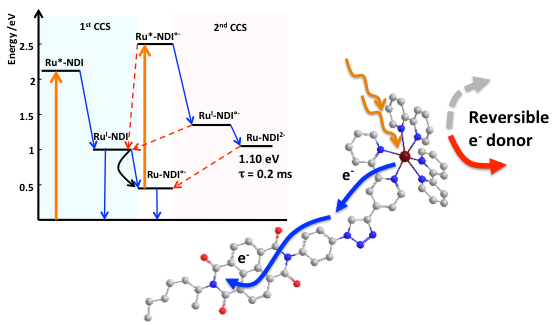
expériences menées par excitations séquentielles résolues en temps ont aussi donné accès à tous les processus de relaxation en compétition (transfert de charge inverse, FRET, désexcitation non-radiative, etc.). Élucider de tels processus intramoléculaires et/ou intermoléculaires est un prérequis incontournable dans la mise au point de systèmes photocatalytiques pour la conversion de l’énergie solaire en carburant moléculaire.
Time-Resolved Interception of Multiple-Charge Accumulation in a Sensitizer–Acceptor Dyad. S. Mendes marinho, M.-H. Ha-Thi, V.-T. Pham, A. Quaranta, T. Pino, C. Lefumeux, T. Chamaillé, W. Leibl, A. Aukauloo (2017) Angew. Chem. Int. Ed. 11, 15936–15940
Spectroscopie d’absorption des rayons-X résolue en temps, technique sophistiquée pour suivre l’accumulation des électrons
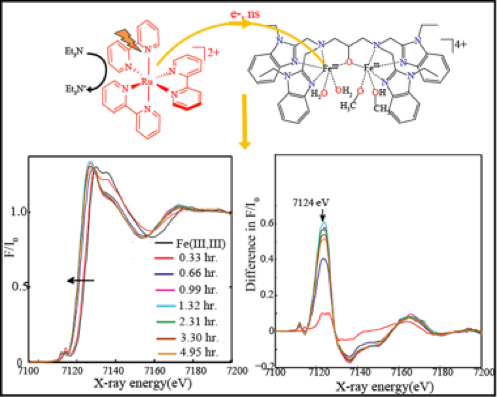
Cette étude fait suite notre publication dans Angewandte Chemie (2015) sur la photocatalyse d’oxydation de substrat organique. En collaboration avec Dr. D. Moonshiram au Argonne National Lab USA, nous avons utilisé la spectroscopie d’absorption des rayons X résolue en temps afin de suivre l’accumulation photoinduite de deux électrons au niveau du catalyseur, un complexe dinucléaire de Fe(III)-Fe(III) en présence d’un donneur d’électron. L’étude d’EXAFS couplée aux calculs DFT nous a permis de mettre en évidence la formation du complexe di-réduit Fe(II)-Fe(II), un intermédiaire essentiel dans notre proposition mécanistique.
Elucidating the light-induced charge accumulation in an artificial analogue of methane monooxygenase enzymes using time-resolved x-ray absorption spectroscopy. D. Moonshiram, A. Picon, Á. Vázquez-Mayagoitia, X. Zhang, M.-F. Tu, P. Garrido-Barros, J.-P. Mahy, F. Avenier, A. Aukauloo (2015) Chem. Commun. 53, 2725-2728
Autre défi
Un problème récurrent dans la recherche menée en photocatalyse multi-électronique pour la transformation de petites molécules (H2O, H2, CO2, CH4, N2…) reste l’utilisation de donneurs/accepteurs sacrificiels d’électrons. L’emploi de ces composants chimiques reste un frein pour le développement des procédés durables. Il est donc urgent d’exclure ces ingrédients dans les processus photocatalytiques. Nous nous attelons à cette tâche avec des collègues de Marseille dans le cadre du projet ANR Multiplet pour la photocatalyse des réactions d’oxydation. Pour ce faire nous mettons en œuvre une Laccase, enzyme capable de consommer des électrons de façon catalytique pour réduire O2 en H2O.
Pour plus de détails, consulter la liste complète des publications ICI.
Dernière mise à jour le 01.08.2023