Les recherches se portent sur
Animatrice : Hafsa Korri-Youssoufi
Animateur : Zakaria Halime
Animateur : David Bonnaffé
Animateur : Philippe Lesot
Animatrice : Dominique Guianvarc’h
L’axe CB2MR regroupe des chercheurs et enseignants-chercheurs aux compétences variées – synthèse, catalyse et analyse – mais dont les thèmes de recherche s’articulent autour des trois piliers que sont la chimie, le vivant et son environnement. L’équipe se structure autour de 5 thématiques. Deux thèmes de recherche sont étroitement liés à la synthèse organique avec le développement de nouvelles méthodologies de synthèse permettant d’accéder à des outils moléculaires innovants pour l’étude de processus biologiques. Ils couvrent les domaines des glycosciences et de la chémo-biologie. Un troisième thème de recherche s’inspire des métalloprotéines pour développer de nouveaux catalyseurs métalliques bioinspirés, soit sous forme de complexes de coordination pour l’activation de petites molécules, soit sous forme d’enzymes artificielles pour des applications thérapeutiques, théranostiques ou environnementales. Enfin, deux thèmes de recherche sont quant à eux orientés vers l’analyse. L’un est dédié au développement de nouvelles méthodologies de spectroscopie RMN, notamment en milieu orienté, pour repousser les limites de l’analyse structurale des molécules en solution. Alors que l’autre concerne le développement de biocapteurs électrochimiques impliquant l’utilisation d’enzymes à leur surface pour des applications médicales et environnementales.
Les membres de l’axe


Les thématiques de recherche
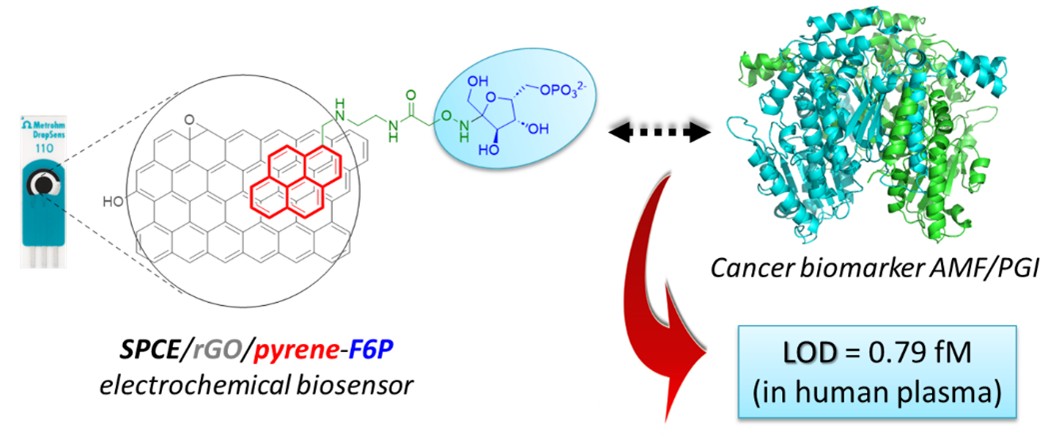
Biocapteurs et inhibiteurs enzymatiques : conception et applications
(H. Dorizon, H. Korri-Youssoufi, L. Salmon, J. Guo)
Les recherches développées au sein de cette thématique associent la chimie bioorganique et organique, l’enzymologie, ainsi que la conception de biocapteurs et de systèmes analytiques. Les objectifs du thème sont de suivre les interactions de reconnaissance moléculaires d’espèces chimiques et biologiques à l’échelle moléculaire ainsi que l’identification de cibles thérapeutiques originales pour le développement de nouveaux médicaments, intégrant des outils de diagnostic et des approches théranostiques.
Notre objectif est, d’une part, l’étude fondamentale de processus biologiques au niveau moléculaire, et d’autre part, la mise au point de molécules et de dispositifs capables de répondre aux grands défis sociétaux liés aux domaines de la santé, de l’agroalimentaire et de l’environnement. Les axes développés sont :
- la synthèse et l’évaluation cinétique d’inhibiteurs d’enzymes d’intérêt, et la résolution des structures cristallographiques des complexes enzyme-inhibiteur associés,
- la compréhension des mécanismes d’action de biomolécules (enzymes, métalloenzymes, cytokines…) et de leurs interactions avec des entités naturelles ou synthétiques (substrats, inhibiteurs, biorécepteurs, toxines, bactéries…),
- la synthèse de macrocycles et de nanomatériaux pour le développement de sondes de reconnaissance optique et électrochimique,
- le développement d’approches innovantes pour la détection en temps réel ultrasensible et spécifique d’analytes d’intérêt (ADN, anticorps, protéines, toxines, métabolites, ions métalliques,…) dans les fluides humains, de polluants dans les matrices environnementales ou pour le contrôle alimentaire, basées sur une lecture directe par voie électrochimique ou optique, sans amplification de l’échantillon,
- l’intégration de biocapteurs dans des microsystèmes, tels que des dispositifs imprimables, portables et flexibles.
Chimie Bioinorganique
Animateur : Zakaria HALIME
(F. Avenier, F. Banse, W. Ghattas, Z. Halime, C. Herrero, R. Ricoux, K. Sénéchal-David, E. Crétal, S. Fatima, T. Lambert, S. Nicolas, L. Tesson, F. Vinchon, A. Vishwakarna, S. Wiorek, A. Patwa, M. Abd el Sater, M. Sacleux)
La thématique Chimie Bioinorganique rassemble des activités centrées sur la conception de systèmes moléculaires inspirés des métalloenzymes. Ces systèmes sont dédiés à l’activation et la transformation de molécules d’intérêt biologique, énergétique, environnemental, ou à la compréhension des mécanismes enzymatiques. Elle couvre la synthèse de complexes métalliques bioinspirés, l’ingénierie de métalloenzymes artificielles ainsi que l’étude de leurs mécanismes. En s’appuyant sur la chimie de coordination, les effets de seconde sphère, l’électro- et la photocatalyse, cette thématique vise à comprendre et reproduire les fonctions catalytiques naturelles (oxydations sélectives, transferts d’électrons ou/et de protons, activation rédox). L’objectif commun est le développement de catalyseurs sélectifs, efficaces et éco-compatibles, utilisables en chimie fine, dépollution ou conversion de l’énergie. En combinant modélisation, biomimétisme et stratégies supramoléculaires innovantes, la thématique propose une vision intégrée et durable de la catalyse bioinspirée. Cette Thématique s’articule autour de 4 sous-thématiques :
Complexes de Fer, catalyse et mécanismes (F. Avenier, F. Banse, Z. Halime, Katell Senechal-David)
Catalyseurs supramoléculaires bioinspirés (W. Ghattas, F. Avenier, R. Ricoux, Katell Senechal-David).
Catalyse bioinspirée pour la conversion d’énergie (Z. Halime, F. Banse, W. Ghattas)
Métalloenzymes artificielles (W. Ghattas, F. Avenier, R. Ricoux)
(F. Avenier, F. Banse, Z. Halime, Katell Senechal-David)
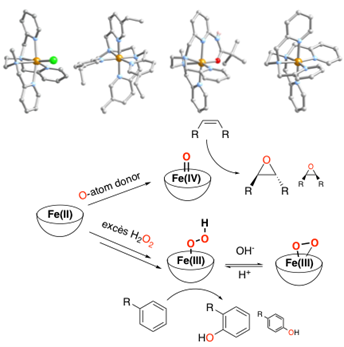
En synthèse chimique, la conversion de molécules organiques en produits oxydés à plus haute valeur ajoutée est souvent effectuée dans des conditions stœchiométriques et sévères (hautes température et pression, oxydants forts générant des sous-produits nocifs ou toxiques). La recherche de solutions alternatives utilisant le dioxygène moléculaire (oxydant très bon marché, abondant et bénin d’un point de vue environnemental) est donc un enjeu évident. Cependant, à cause de son état fondamental triplet de spin (S=1), la réaction de O2 avec des espèces diamagnétiques est infiniment lente. Son activation est donc nécessaire afin de franchir la barrière cinétique.
De manière intéressante, certaines métalloenzymes (monooxygénases, dioxygénases, hydroxylases) sont capables d’utiliser O2 pour catalyser l’oxydation chemo- et régiosélective de petites molécules organiques dans des conditions douces. Pour cela, elles réalisent « l’activation réductrice du dioxygène » dans leur site actif qui contient un ion métallique biodisponible et abondant (Fe, Mn, Cu…) : l’activation de O2 nécessite sa réduction partielle et contrôlée. La modélisation de ces catalyseurs naturels par des systèmes synthétiques plus simples paraît donc pertinente tant d’un point de vue fondamental (détermination des mécanismes réactionnels) que pratique (valorisation de matière première abondante).
Avec des systèmes synthétiques peu coûteux (complexes de FeII non hémiques) et en présence d’oxydants chimiques, il est possible de générer des intermédiaires réactionnels Fe-peroxo, Fe-oxo de haute valence, capables d’oxyder de petites molécules organiques. Ces espèces sont analogues à celles identifiées dans le cycle catalytique des enzymes. Nos objectifs sont :
1/ de comprendre la réactivité intrinsèque de ces espèces afin de les exploiter en catalyse (Isolement et étude des intermédiaires réactionnels) ;
2/ de reproduire plus fidèlement la réactivité des systèmes naturels en utilisant des petites molécules renouvelables (O2, H2O) plutôt que des oxydants chimiques (Activation de petites molécules) ;
3/ d’améliorer la réactivité de ces catalyseurs en jouant sur leur seconde sphère de coordination via des effets supramoléculaires (Insertion des catalyseurs dans des systèmes supramoléculaires et effets de seconde sphère).
Dans un contexte plus large de transition énergétique, notre équipe s’investit dans le développement de systèmes catalytiques capables de stocker efficacement l’énergie sous forme de liaisons chimiques. En nous inspirant des sites actifs de certaines métalloenzymes, nous cherchons à concevoir des catalyseurs moléculaires performants pour l’activation et la transformation de petites molécules d’intérêt énergétique. Plus particulièrement, nos travaux portent sur des transformations électro- et photocatalytiques pour la réduction du CO₂, la réduction de l’O₂, la production de d’hydrogène ainsi que l’oxydation de l’eau. L’objectif est de comprendre et de maîtriser les paramètres structuraux gouvernant ces transformations afin de proposer des solutions durables et innovantes pour la conversion et le stockage de l’énergie.
Exemples d’études récentes :
Hinged Carboxylate in the Artificial Distal Pocket of an Iron Porphyrin Enhances CO2 Electroreduction at Low Overpotential. Advanced Science (2025).
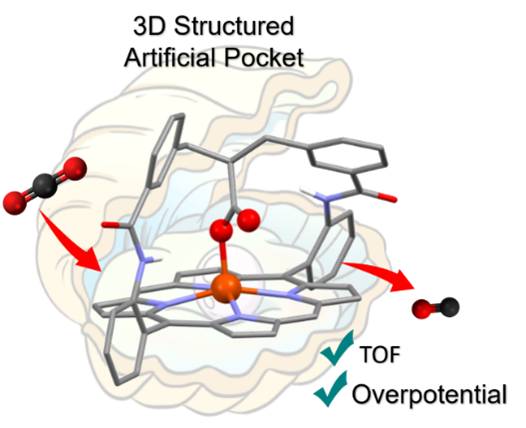
To efficiently capture, activate, and transform small molecules, metalloenzymes have evolved to integrate a well-organized pocket around the active metal center. Within this cavity, second coordination sphere functionalities are precisely positioned to optimize the rate, selectivity, and energy cost of catalytic reactions. Inspired by this strategy, an artificial distal pocket defined by a preorganized 3D strap was introduced on an iron-porphyrin catalyst (sc-Fe) for the CO2-to-CO electrocatalytic reduction. Combined electrochemical, kinetic, and computational studies demonstrate that the adequate positioning of a carboxylate/carboxylic group acting in synergy with a trapped water molecule within this distal pocket remarkably enhances the reaction turnover frequency (TOF) by four orders of magnitude compared to the perfluorinated iron-tetraphenylporphyrin catalyst (F20Fe) operating at a similar low overpotential. A proton-coupled electron transfer (PCET) was found to be the key process responsible for the unexpected protonation of the coordinating carboxylate, which, upon CO2 insertion, shifts from the first to the second coordination sphere to play a possible secondary role as a proton relay.
Time-Resolved Mechanistic Depiction of Photoinduced CO2 Reduction Catalysis on a Urea-Modified Iron Porphyrin. Angew. Chem. Int. Ed., e202407723
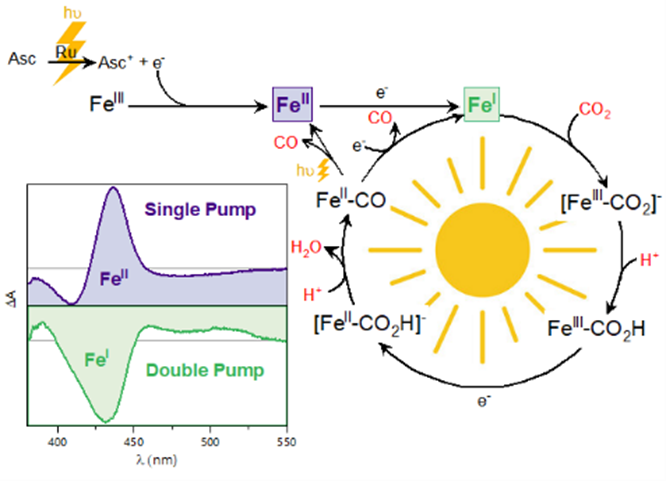
In spite of the advancements in homogeneous catalysis, the development of the next generation of catalysts requires a complete understanding of the fundamental photoinduced processes taking place prior to and after activation of the substrate by the catalyst. In this work, we employ a state-of-the-art nanosecond optical transient absorption spectroscopic setup with a double excitation capability to induce charge accumulation and trigger the reduction of CO2 to carbon monoxide (CO). Our biomimetic system is composed of a urea-modified iron(III) tetraphenylporphyrin (UrFe(III)) catalyst, the prototypical [Ru(bpy)3]2+ (bpy=2,2’-bipyridine) used as a photosensitizer, and sodium ascorbate as an electron donor. Under inert atmosphere, we show that two electrons can be successively accumulated on the catalyst as the fates of the photogenerated UrFe(II) and UrFe(I) reduced species are tracked. In the presence of CO2, the catalytic cycle is kick-started providing further evidence on CO2 activation by the UrFe catalyst in its formal Fe(I) oxidation state.
Second Coordination Sphere Effect Shifts CO2 to CO Reduction by Iron Porphyrin froFe0 to FeI. Angew. Chem. Int. Ed. Engl., e202314439
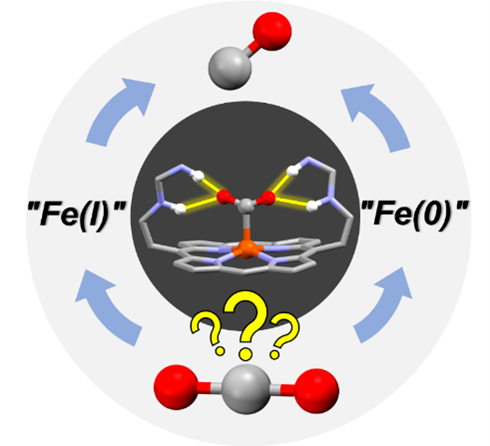
Iron porphyrins are among the most studied molecular catalysts for carbon dioxide (CO2) reduction and their reactivity is constantly being enhanced through the implementation of chemical functionalities in the second coordination sphere inspired by the active sites of enzymes. In this study, we were intrigued to observe that a multipoint hydrogen bonding scheme provided by embarked urea groups could also shift the redox activation step of CO2 from the well-admitted Fe(0) to the Fe(I) state. Using EPR, resonance Raman, IR and UV-Visible spectroscopies, we underpinned a two-electron activation step of CO2 starting from the Fe(I) oxidation state to form, after protonation, an Fe(III)‒COOH species. The addition of another electron and a proton to the latter species converged to the cleavage of a C‒O bond with the loss of water molecule resulting in an Fe(II)‒CO species. The DFT analyses of these postulated intermediates is in good agreement with our collected spectroscopic data, thus allowing us to propose an alternative pathway in the catalytic CO2 reduction with iron porphyrin catalyst. Such a remarkable shift opens new lines of research in the design of molecular catalysts to reach low overpotentials in performing multi-electronic CO2 reduction catalysis.
Unlocking Full and Fast Conversion in Photocatalytic Carbon Dioxide Reduction: Applications in Radio-Carbonylation. Nat. Commun. 14, 4451-2373
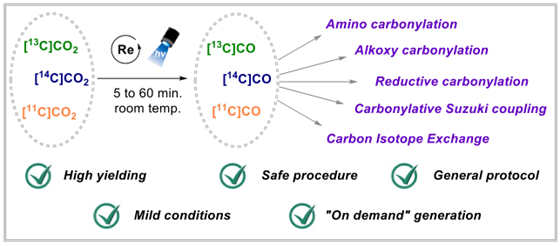
Harvesting sunlight to drive carbon dioxide (CO2) valorisation represents an ideal concept to support a sustainable and carbon-neutral economy. While the photochemical reduction of CO2 to carbon monoxide (CO) has emerged as a hot research topic, the full CO2 -to-CO conversion remains an often-overlooked criterion that prevents a productive and direct valorisation of CO into high-value-added chemicals. Herein, we report a photocatalytic process that unlocks full and fast CO2-to-CO conversion (< 10 minutes) and its straightforward valorisation into human health related field of radiochemistry with carbon isotopes. Guided by reaction-model-based kinetic simulations to rationalize reaction optimisations, this manifold opens new opportunities for the direct access to 11C- and 14C-labeled pharmaceuticals from their primary isotopic sources [11C]CO2 and [14C]CO2.
Efficient Hydrogen Production at pH 7 in Water with a Heterogeneous Electrocatalyst Based on a Neutral Dimeric Cobalt-Dithiolene Complex. ACS Catalysis 13, 2367-2373
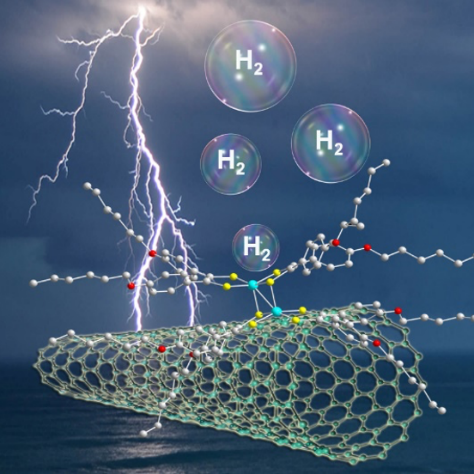
The development of efficient hydrogen production technologies is fundamental for global sustainability. As such, electrocatalysts derived from earth abundant metal complexes are appealing, and interesting performances have typically been disclosed under acidic conditions in organic solvents. However, their applicability under relevant pH neutral conditions is underexplored. Herein, we demonstrate that non-ionic, dimeric cobalt-dithiolene complexes supported on multi-wall carbon nanotubes (MWCNTs)/carbon paper (CP) electrode are powerful electrocatalysts for hydrogen production in aqueous media at pH 7. The high turnover numbers encountered (TON up to 50980) after long reaction times (up to 16 hours) are reasoned by the increased electro-active cobalt concentration on the modified electrode, which is ca. 4 times higher than that of a state-of-the-art cobalt porphyrin electrocatalyst. These findings point out that immobilizing well-defined, multinuclear low-cost metal complexes on carbon material is a promising strategy to design highly electroactive electrodes.
Bio-Inspired Bimetallic Cooperativity Through a Hydrogen Bonding Spacer in CO2 Reduction. Angew. Chem. Int. Ed. 62, e202214665
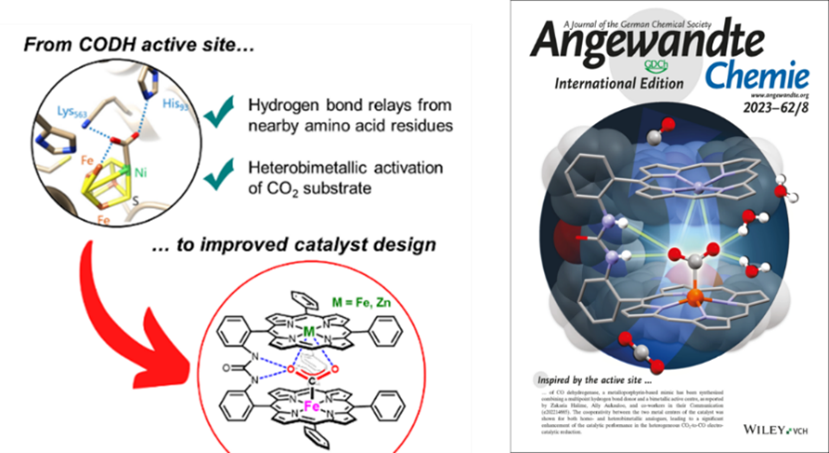
At the core of carbon monoxide dehydrogenase (CODH) active site two metal ions together with hydrogen bonding scheme from amino acids orchestrate the interconversion between CO2 and CO. We have designed a molecular catalyst implementing a bimetallic iron complex with an embarked second coordination sphere with multi-point hydrogen-bonding interactions. We found that, when immobilized on carbon paper electrode, the dinuclear catalyst enhances up to four-fold the heterogeneous CO2 reduction to CO in water with an improved selectivity and stability compared to the mononuclear analogue. Interestingly, quasi-identical catalytic performances are obtained when one of the two iron centers was replaced by a redox inactive Zn metal, questioning the cooperative action of the two metals. Snapshots of X-ray structures indicate that the two metalloporphyrin units tethered by a urea group is a good compromise between rigidity and flexibility to accommodate CO2 capture, activation, and reduction.
Couvertures :

Métalloenzymes artificielles
(W. Ghattas, F. Avenier, R. Ricoux)
Les métalloenzymes artificielles sont des catalyseurs hybrides conçus en laboratoire, résultant de l’intégration d’un site métallique de synthèse au sein d’une macromolécule protéique ou d’un support biologique, et capables de catalyser des réactions chimiques parfois inédites en biologie. Elles peuvent être préparées non seulement à partir d’enzymes existantes dont l’activité est modifiée ou redirigée, mais aussi à partir de protéines dépourvues d’activité catalytique native ou de structures conçues de novo, en y introduisant des sites de coordination adaptés. Ces approches tirent parti des progrès récents en génie des protéines, en conception de sites métalliques et en évolution dirigée. En suivant ces approches, nous préparons des métalloenzymes artificielles pour des applications variées ; les exemples les plus récents de notre équipe sont présentés ci-dessous.
Binding and Stabilization of a Semiquinone Radical by an artificial Metalloenzyme containing a Binuclear copper (II) cofactor. ChemBioChem e202400139
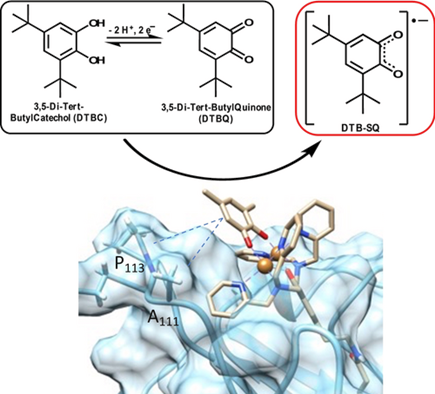
We aimed at the preparation of an artificial metalloenzyme that catalyze catechol oxidation. For that, a binuclear Cu(II) cofactor known to catalyze this reaction was covalently bound to a lauric acid anchor. By following the so-called “Trojan horse” strategy, we combined the resulting conjugate with beta-lactoglobulin (βLG) to generate a new biohybrid owing to the affinity of this protein to fatty acids. This biohybrid was examined for its effectiveness in the oxidation of a catechol derivative to the corresponding quinone. The resulting biohybrid did not exhibit the sought after catecholase activity, likely due to its ability to bind and stabilize the semiquinone radical intermediate DTB-SQ. This semi-quinone radical was stabilized only in the presence of the protein and was characterized using optical and magnetic spectroscopic techniques, demonstrating stability for over 16 hours. Molecular docking studies revealed that this stabilization could occur owing to interactions of the semi-quinone with hydrophobic amino acid residues of βLG.
An artificial metalloprotein with metal-adaptive coordination sites and Ni-dependent quercetinase activity. J. Inorg. Biochem. 10.1016/j.jinorgbio.2022.111914
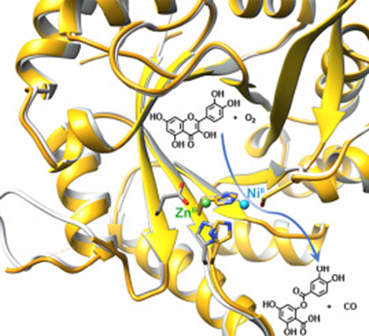
Engineering non-native metal active sites into proteins using canonical amino acids offers many advantages for catalysis but is hampered by significant challenges. The TIM barrel protein, imidazole glycerol phosphate synthase from the hyperthermophilic organism Thermotoga maritima (tHisF), is well-suited for the construction of artificial metalloenzymes by this approach. To this end, we have generated a tHisF variant (tHisFEHH) with a Glu/His/His motif for metal ion coordination. Crystal structures of ZnII:tHisFEHH and NiII:tHisFEHH reveal that both metal ions bind to the engineered histidines. However, the two metals bind at distinct sites with different geometries, demonstrating the adaptability of tHisF. Only ZnII additionally ligates the Glu residue and adopts a tetrahedral geometry. The pseudo-octahedral NiII site comprises the two His and a native Ser residue. NiII:tHisFEHH catalyzes the oxidative cleavage of the flavanols quercetin and myricetin, providing an unprecedented example of an artificial metalloprotein with quercetinase activity.
(W. Ghattas, F. Avenier, R. Ricoux, Katell Senechal-David)
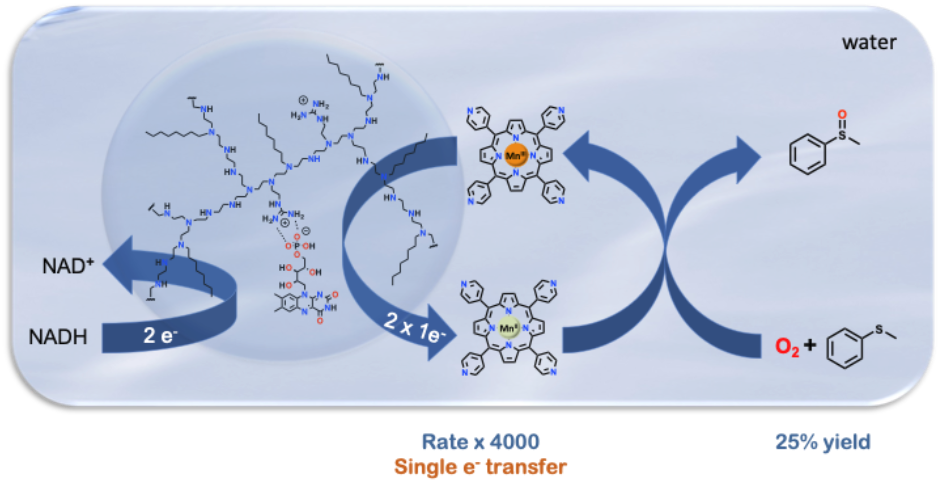
Les métalloenzymes, ou les enzymes d’une façon plus générale, exploitent la structure tridimensionnelle des protéines pour confiner leurs cofacteurs dans un environnement hydrophobe localement polarisé, leur conférant ainsi des réactivités différentes de celle opérant simplement en solution aqueuse. Dans ce contexte, nous développons des catalyseurs supramoléculaires qui reprennent ce principe en incorporant des cofacteurs flavinique au sein d’un polymère hydrosoluble, mais dont le microenvironnement est localement hydrophobe. Cela nous permet notamment de développer des réductases artificielles capables de collecter les électrons de NADH et de les transférer à un centre métallique comme le font les réductases naturelles pour activer le dioxygène, ou encore de catalyser la réaction de Baeyer-Villiger dans l’eau comme le font les Baeyer-Villiger monoxygénases naturelles.
Glycochimie
(L.-A. Barel, J. Hénault, C. Le Narvor, G. Blondy, U. Koty, Q. Lin)
We develop new methodologies in organic synthesis as key steps to prepare new chemical-biology tools designed to understand, at the molecular level, biological processes involving carbohydrates. Our expertise in glycochemistry, including chemical and chemoenzymatic oligosaccharide synthesis, (bio)orthogonal conjugation strategies and supramolecular chemistry gives a unique edge to our research and a solid base to our ambition to bring innovative solution for human health.
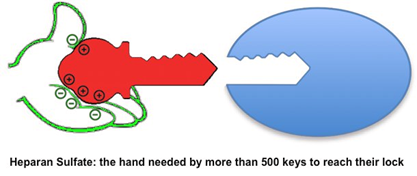
We concentrate on Glycosaminoglycans (GAGs), especially Heparan Sulfate (HS), which are linear and sulfated polysaccharides able to modulate the activities of more than 500 extracellular proteins, including established therapeutic targets. Behaving like a hand needed by Heparan Sulfate Binding Proteins (HSBP) to reach their lock, HS chains are nowadays recognized as crucial additional partners in the « Lock and key » paradigm.
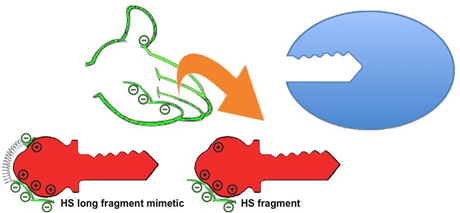
A tightly regulated biosynthesis allows generating several levels of molecular diversity and encoding protein-specific sequences depending on cell type or activation. Disrupting such selective interactions represents a promising field that could advantageously complement current therapies and that is thoroughly explored by our group, with an emphasis on HIV gp120 and cytokines and chemokines involved in immune and inflammatory diseases and cancers.
Towards this goal we develop new methodologies in organic chemistry and integrate different chemical biology approaches into a global toolbox, whose aim is to allow the rational design and systematic optimization of glycoligands able to disrupt a physiologically relevant interaction between a protein of therapeutic interest and HS chains.
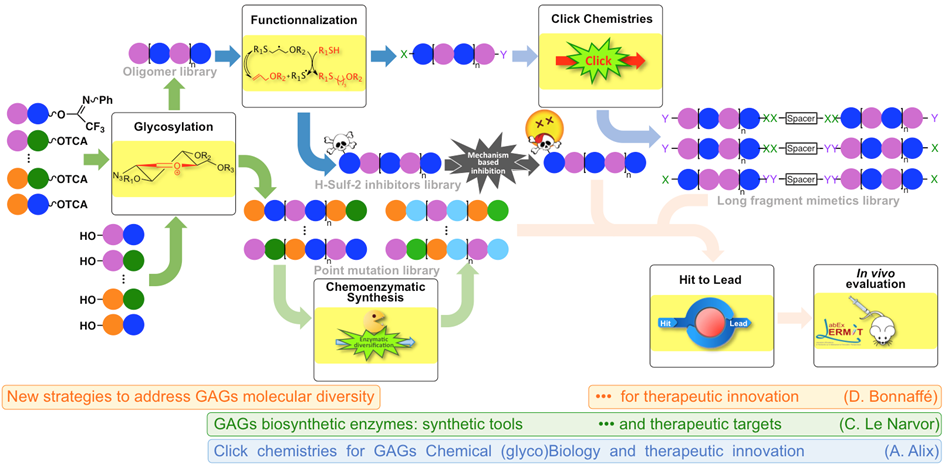
The projects developed in the Glycosaminoglycans and Molecular Diversity group are deeply interwined and benefit from our recognized expertise to integrate into coherent and coordinated approaches:
•New strategies to address GAGs molecular diversity for therapeutic applications
•GAGs biosynthetic enzymes as synthetic tools and therapeutic targets
•Click chemistries for GAGs chemical (glyco)biology and therapeutic innovation
Développements méthodologiques et analytiques en RMN isotrope et anisotrope
Animateur : P. Lesot
(C. Aroulanda, P. Berdagué, B. Gouilleux, P. Lesot, A. Meddour, D. Merlet, F.-M. Moussallieh, B. Rousseau, V. Ciapolino)
Description de la thématique RMN :
La thématique RMN de l’axe CB2MR a pour objet le développement de méthodes de spectroscopie RMN liquide et en milieux anisotropes comprenant : la préparation d’échantillon, la conception de séquence d’impulsions, le traitement des données et le développement d’outils de modélisation moléculaires. Ces travaux méthodologiques en chimie structurale visent à répondre à diverses questions et besoins en chimie organique et bio-organique. Une attention particulière est accordée à l’utilisation de solvants orientés chiraux, tels que les cristaux liquides lyotropes, préparés à partir de polymères hélicoïdaux polypeptidiques et polyacétylèniques. Les expériences de RMN effectuées dans de tels milieux orientés permettent d’observer et de mesurer des interactions RMN anisotropes résiduelles comme le couplage dipolaire (RDC), le couplage quadrupolaire (RQC) et l’anisotropie de déplacement chimique (RCSA), tout en gardant une haute résolution. Ces interactions anisotropes, non mesurables dans les liquides isotropes, sont riches en informations moléculaires et structurales, en particulier du point de vue stéréochimique, ce qui est à la base de quatre sous-thématiques décrites ci-dessous.

Personnels permanents (ordre alphabétique) (en 2026) :
C. Aroulanda, Ph. Berdagué, B. Gouilleux, Ph. Lesot, A. Meddour, D. Merlet, F.-M. Moussallieh, B. Rousseau.
Personnels non permanents (en 2026) :
V. Chiapolino (Thèse)
C. Aroulanda, P. Berdagué, V. Chiapolino, B. Gouilleux, P. Lesot, A. Meddour, F.-M. Moussallieh.
Lorsqu’une molécule chirale est dissoute dans un cristal liquide chiral, ses énantiomères peuvent adopter une orientation moyenne différente par rapport au champ magnétique B0 du spectromètre RMN. Cela conduit à des interactions RMN anisotropes dont les observables résiduelles (RDC, RQC et RCSA) varient d’un énantiomère à l’autre, ce qui permet une discrimination spectrale de l’ensemble des stéréoisomères. Tout noyau magnétiquement actif en RMN est une sonde potentielle de détection spectrale de la discrimination chirale.
Cette sous-thématique se développe sur divers aspects de la discrimination chirale par RMN multinucléaire. Elle se concentre actuellement sur l’analyse énantiomérique de principes actifs chiraux fluorés par RMN 19F en solvant orienté où la résolution énantiomérique peut se faire ici par variation de RCSA(19F) et de RDC(19F-19F). Ces travaux s’effectuent actuellement dans le cadre du projet ANR INENDRA (https://anr.fr/Projet-ANR-23-CE29-0005).
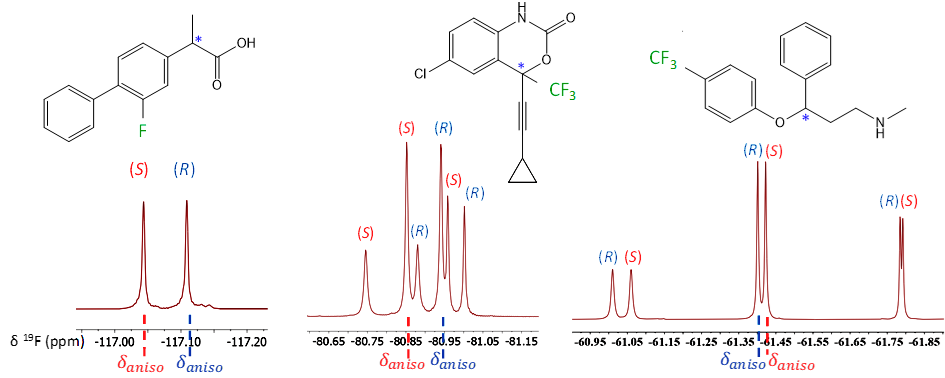
Figure : Exemples de discrimination énantiomérique de principes actifs chiraux par RMN 1D 19F-{1H} en milieu orientant du type poly-g-benzyl-L-glutamate (PBLG)/CHCl3. De gauche à droite : Flurbiprofène, Efavirenz et Fluoxétine.
C. Aroulanda, P. Berdagué, B. Gouilleux, P. Lesot, A. Meddour, B. Rousseau.
Les interactions RMN anisotropes résiduelles (RCSA(13C), RDC(13C-1H) ou RQC(2H)) encodent une information géométrique (3D) et configurationnelle. Leur exploitation offre donc des informations structurales importantes, généralement complémentaires aux informations de couplages scalaires J ou associées aux effets NOE, mesurés en RMN isotrope. Elles sont aussi porteuses d’informations moléculaires de type isotopique (2H/1H, 13C/12C). Autour des noyaux deutérium sont développés depuis plusieurs années des outils robustes dédiés à la mesure et à l’exploitation analytique des couplages quadrupolaires résiduels deutérium (RQC(2H)) sur des molécules marquées spécifiquement (100%) ou en abondance naturelle (DANA) (1,55 10-2%). Ces outils permettent l’analyse chirale et/ou prochirale, la détermination du profil isotopique 2H/1H de molécules naturelles et plus récemment sont utilisés pour la détermination configurationnelle de produits naturels chiraux (bioactifs).
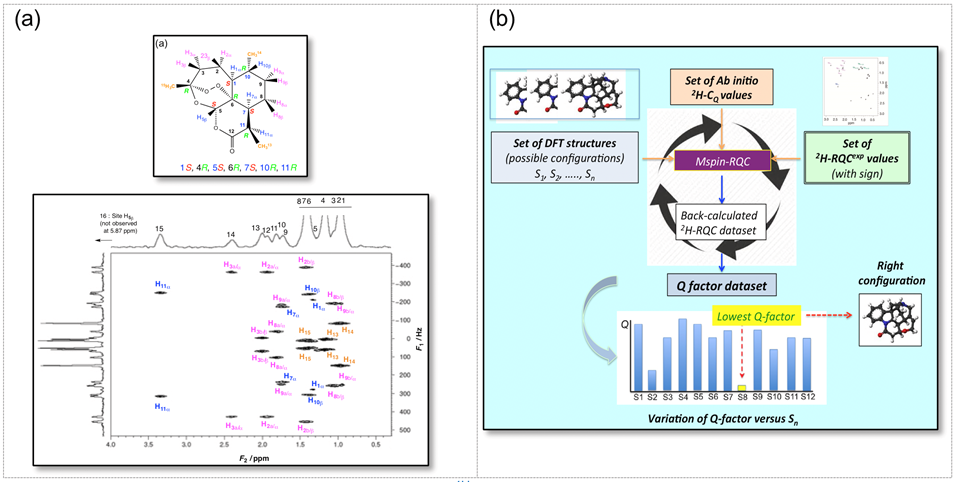
Figure : (a)Exemple de spectre 2D QUOSY DANA-{1H} de l’Artémisinine. (b) Principe simplifié de l’analyse de la configuration relative par RMN 2D DANA.
C. Aroulanda, B. Gouilleux, P. Lesot, A. Meddour, D. Merlet.
Dans cette sous-thématique sont explorées et développées de nouvelles approches méthodologiques RMN, impliquant l’utilisation de systèmes (chiraux ou pas) isotropes, anisotropes, biphasiques ou bimésophasiques un nouveau concept d’échantillons RMN orientés récemment proposés à l’ICMMO et qui présentent simultanément deux types de domaines orientants après leur démixtion forcée (par centrifugation) ou naturelle (par gravitation).
Ces nouvelles approches se déclinent sous différents aspects, mais reposent en particulier sur la conception de séquences RMN multidimensionnelle (nD) et multi-impulsionnelles innovantes (milieux isotrope et anisotrope). On pourra ainsi citer la mise au point de séquences RMN 1D/2D incluant : i) la saturation sélective (ou multi-sélective) de signaux RMN indésirables, ii) les expériences 2D avec encodage en fréquence G-SERF (Gradient-based SElective ReFocalisation) et UF (Ultrafast), iii) les expériences 1D STD (Saturation-Transfert-Difference), iv) les approches de type « OVS » (Outer Volume Suppression) pour l’enregistrement de spectres spatialement localisés, v) les expériences 2D basées sur une acquisition non-uniforme des données expérimentales (NUS), et enfin vi) leurs diverses combinaisons. Plus récemment, l’analyse RMN en milieu anisotrope impliquant des spectromètres RMN dits « bas-champ » (Benchtop NMR) a été initiée et les résultats sont prometteurs.
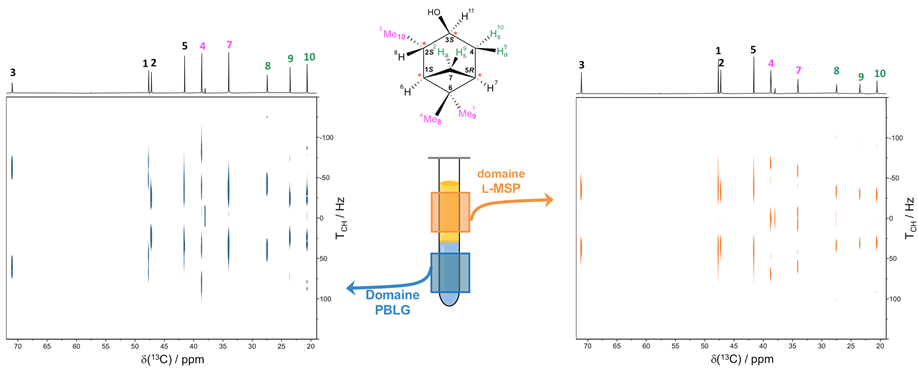
Figure : Spectres 2D OVS TCH-Résolue 13C enregistrés sur les deux domaines polymériques d’un système bimésophasique chiral « PBLG/L-MSP » : (à droite) PBLG, (à gauche) L-MSP.
C. Aroulanda, P. Berdagué, B. Gouilleux, P. Lesot, F.-M. Moussallieh, B. Rousseau.
Les mécanismes de reconnaissance chirale et d’orientation moléculaire moyenne de solutés au sein des cristaux liquides lyotropes à base de polymères hélicoïdaux (par exemple le PBLG) sont des phénomènes complexes (généralement associés à la conformation des chaines latérales du polymère) et ne sont toujours pas bien compris et donc bien maitrisés, aujourd’hui. Une meilleure compréhension de ces phénomènes devrait permettre de mieux les décrire. La possibilité de prédire ces phénomènes serait un atout majeur pour, par exemple : i) l’analyse conformationnelle en milieu anisotrope, ii) l’optimisation des conditions expérimentales pour la discrimination énantiomérique, iii) la détermination de la configuration absolue de molécules chirales.
Dans cette sous-thématique, des travaux expérimentaux utilisant des expériences de RMN par STD 1H pour évaluer les interactions « soluté-solvant », des études comparant les caractéristiques orientationnelles de solutés dans différentes mésophases chirales, et des études plus théoriques impliquant de la modélisation moléculaire de ces systèmes orientés/orientants chiraux par dynamique moléculaire (DM) sont menées.
On utilise ces outils de modélisation pour étudier les interactions entre un milieu orientant et son environnement direct en solution. Les premiers travaux ont porté essentiellement sur le polymère chiral PBLG. On étudie l’influence des paramètres de champ de force sur la structure de l’hélice alpha du PBLG, ainsi que l’espace conformationnel visité par la chaine latérale. Ces études sont réalisées dans différents solvants organiques, achiraux, prochiraux, chiraux afin d’évaluer la capacité prédictive de la modélisation moléculaire à reproduire les effets anisotropes (par exemple, les éclatements quadrupolaires) observés expérimentalement.
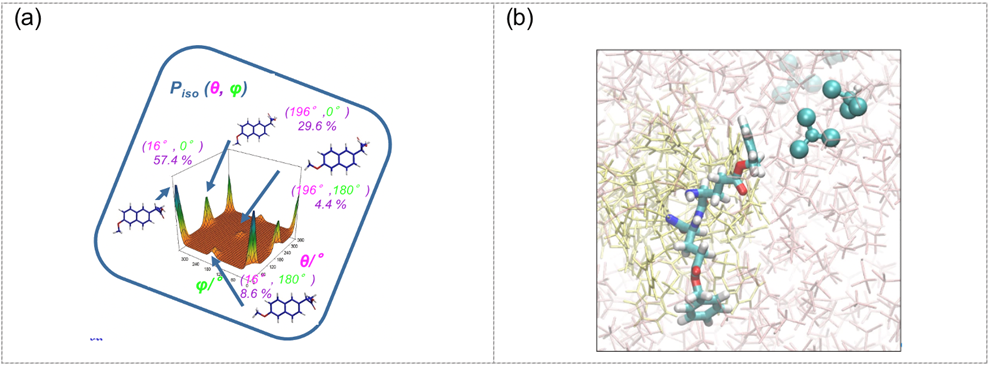
Figure : (a) Distribution des populations conformationnelles du Naproxène en fonction de deux angles de torsion. (b) Représentation du PBLG en solution dans le chloroforme où deux chaînes latérales du PBLG et deux molécules de solvant (chloroforme) sont mises en évidence.
Synthèse de Biomolécules
Animatrice : Dominique Guianvarc’h
(Y. Bourdreux, G. Doisneau, D. Guianvarc’h, D. Urban, D. Ait-Ouarab, C. David, S. Depienne, L. Madegard)

Le thème « Synthèse de Biomolécules » vise à développer de nouvelles méthodologies de synthèse pour créer des outils chimiques innovants permettant d’étudier ou de réguler les processus biologiques à l’échelle moléculaire. Notre groupe réunit des expertises très complémentaires en synthèse organique (synthèse de molécules bioactives, glycochimie, chimie des acides nucléiques, chimie du bore, sondes fluorescentes) et en biochimie (enzymologie, protéines et acides nucléiques). Nous nous intéressons particulièrement à la conception de nouveaux outils chimiques permettant une meilleure compréhension des mécanismes biologiques essentiels, et facilitant leur analyse ou leur modulation avec des applications potentielles dans le domaine de la santé.
Responsable : Yann Bourdreux
Participants : Dominique Guianvarc’h, Gilles Doisneau, Dominique Urban, Léa Madegard, Doria Ait-Ouarab
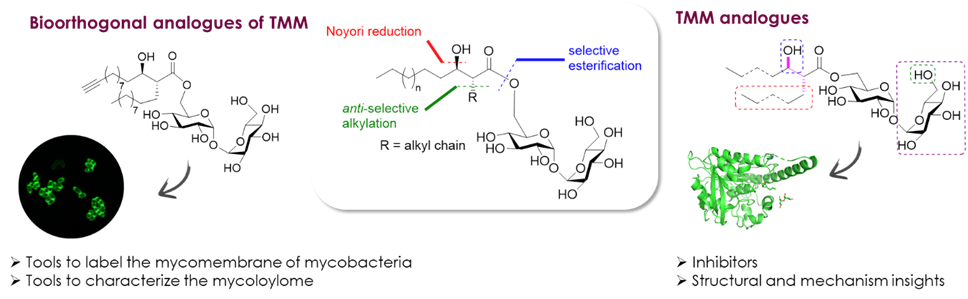
Les mycobactéries, qui comptent notamment l’agent pathogène responsable de la tuberculose, possèdent une membrane particulière, riche en glycolipides complexes dérivés du tréhalose. Forts de notre expertise en chimie du tréhalose et en synthèse d’acides gras complexes, nous développons des outils chimiques innovants pour sonder la mycomembrane. Nos travaux portent sur la conception de marqueurs bioorthogonaux et de sondes fluorescentes à base de tréhalose pour le marquage métabolique, permettant l’imagerie de la paroi bactérienne, l’étude protéomique et l’étude des enzymes impliquées dans la biogenèse de la mycomembrane.
Responsable : Dominique Urban
Participants : Dominique Guianvarc’h, Gilles Doisneau, Yann Bourdreux, Claudia Köhler
Les espèces réactives de l’oxygène (ROS), produites lors d’un stress oxydatif, peuvent induire des dommages moléculaires importants chez les organismes vivants. Nos recherches portent sur le développement de stratégies de synthèse efficaces, l’étude physico-chimique et l’évaluation biologique de nouvelles sondes fluorogéniques « off-on » hautement spécifiques et sensibles pour la détection d’une espèce réactive de l’oxygène, le peroxyde d’hydrogène (H₂O₂). Ces sondes sont basées sur la réactivité accrue des acides boriniques par rapport aux sondes acides boroniques généralement utilisées. Ces déclencheurs hautement réactifs constituent désormais des outils moléculaires polyvalents et sont exploités dans plusieurs projets en cours, ouvrant ainsi de nouvelles perspectives pour des applications théranostiques.

Responsables : Gilles Doisneau, Dominique Guianvarc’h
Participants : Dominique Urban, Yann Bourdreux, Charlène David
La régulation épigénétique de la chromatine bénéficie de plus en plus des approches de chémobiologie, permettant une étude précise des modifications chimiques des protéines et de l’ADN et ouvrant de nouvelles perspectives pour l’innovation thérapeutique.
L’un de nos projets vise à développer des sondes chimiques pour étudier les modifications atypiques de l’ADN présentes chez les parasites responsables de maladies tropicales. En particulier, le β-D-glucosyl-5-hydroxyméthyluracile (base J) est une base d’ADN hypermodifiée, essentielle à la survie du parasite et une cible thérapeutique prometteuse. Nous avons mis au point des voies de synthèse pour la base J et ses analogues, dont plusieurs présentent une activité antiparasitaire.
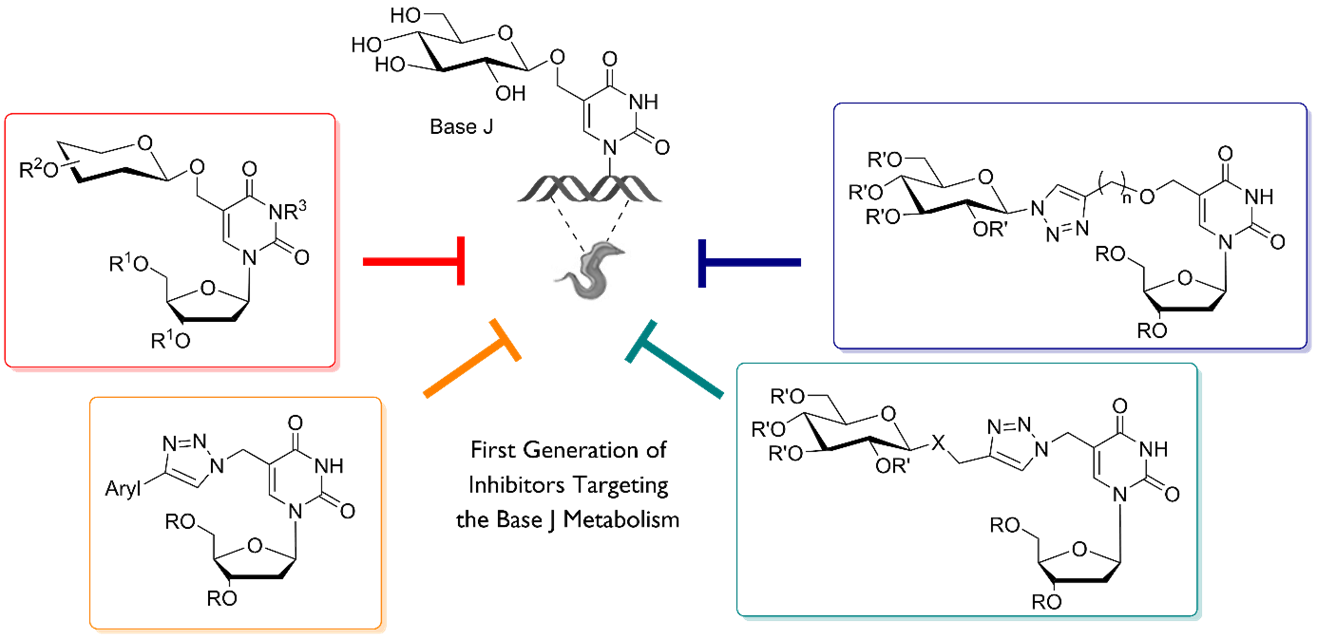
Responsable : Dominique Guianvarc’h
Participants : Dominique Urban, Yann Bourdreux, Gilles Doisneau, Sébastien Depienne
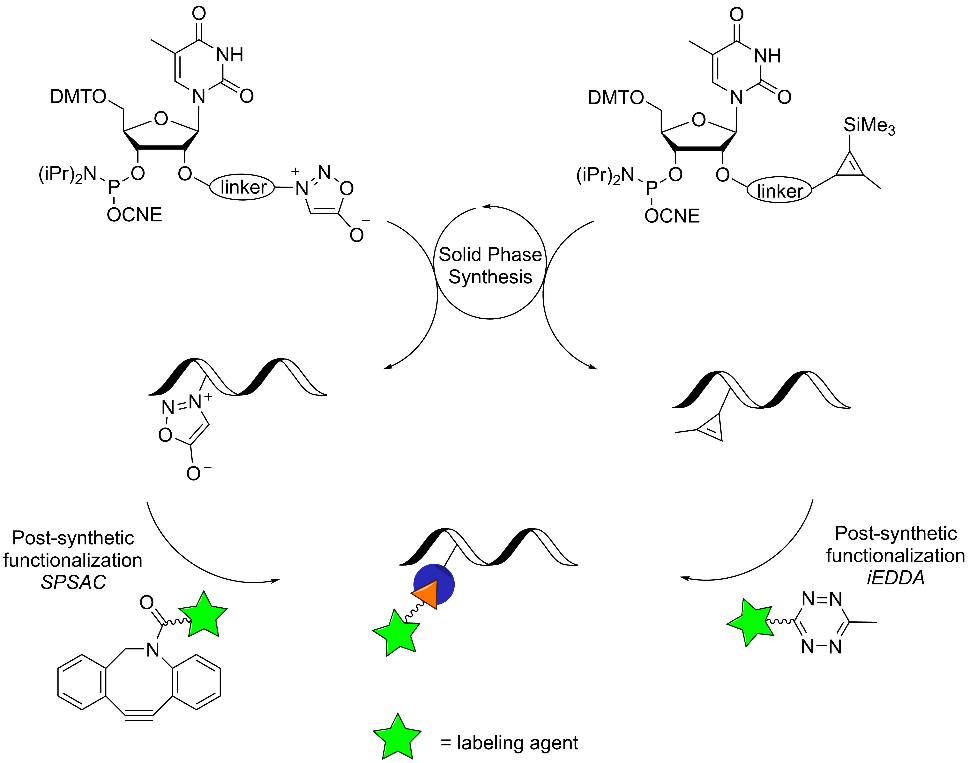
Le développement de méthodes efficaces de conjugaison de sondes, de ligands ou d’agents de vectorisation sur des oligonucléotides (ON) est essentiel tant pour les études fondamentales de chémobiologie que pour l’amélioration des propriétés pharmacologiques des ON en chimie médicinale. En collaboration avec la société Servier, nous avons conçu de nouveaux phosphoramidites de nucléosides fonctionnalisés portant des groupements bioorthogonaux (méthylcyclopropène ou sydnone) permettant le marquage post-synthétique d’ON via des réactions de Diels-Alder à demande électronique inverse (iEDDA), ou par cycloaddition de sydnone-alcyne contraint (SPSAC). Ces nouveaux outils chimiques sont actuellement appliqués à l’étude des oligonucléotides thérapeutiques, ouvrant la voie à un suivi plus précis, à une analyse fonctionnelle et à une optimisation des médicaments de nouvelle génération à base d’acides nucléiques.