
Approche bottom-up (à gauche) et top-down (à droite)
Comment passer d'un modèle énergétique complexe fondé sur la structure éléctronique d'un alliage à un modèle énergétique simple permettant d'en explorer la thermodynamique et d'en analyser les forces motrices ?
Une approche classique consiste à calculer les énergies associées aux interactions locales entre atomes plus proches voisins de structures ordonnées (Cluster Expansion Method). Ces énergies sont ensuite introduites dans des simulations de type Monte Carlo sur réseau. Il s'agit là d'une approche bottom-up : les données obtenues sur les structures basses températures sont utilisées pour décrire les structures hautes températures. Cette méthodologie est efficace mais elle ne permet pas d'extraire les effets chimiques des effets élastiques et ne décrit pas bien la solution solide aléatoire.
On développe une approche top-down qui consiste à décrire la solution solide aléatoire (structure haute température) pour étudier les structures basses températures. Partant d'un potentiel interatomique à N-corps, on détermine les énergies des atomes A et B de l'alliage AcB1-c selon leurs différents environnements locaux et selon la concentration nominale. Pour cela, on considère tous les environnements possibles en proches voisins d'un atome donné, le reste de la boîte étant dans des configurations chimiquement désordonnées de concentration fixée. Toutes les énergies sont obtenues après relaxation des positions atomiques. Nous avons développé un Hamiltonien fondé sur ces énergies de site, capable d'extraire les effets chimiques et élastiques même lorsque ces effets rentrent en compétition.
Proposition de stage Informatique/Physique-Chimie sur le développement d'un code parallélisé intégrant la détermination des énergies de site et leur intégration dans un code Monte Carlo (contacts F. Berthier, J. Creuze)
Proposition de stage/thèse sur le développement de cette approche avec la prise en compte des interactions complexes (contacts F. Berthier, J. Creuze en collaboration avec F. Soisson du CEA/SRMP)
Proposition de stage sur l'application de cette approche à l'étude d'oxydes non-stoechiométriques (contacts F. Berthier, R.Tétot, J. Creuze)
One-Year postdoctoral Fellowship : Sensitivity analysis and learning techniques for mapping the thermodynamic properties of alloys (contact F. Berthier)
Analysis of Au-Pd Driving forces via the Effective Site Energy Model: LRO, Antisites and Enthalpy of Permutation. F. Berthier, B. Legrand, J. Phys.: Condens. Matter, 2020
Order-disorder or phase-separation transition: Analysis of the Au-Pd system by the effective site energy model. F. Berthier, J. Creuze, T. Gabard, B. Legrand, M.-C. Marinica, C. Mottet, Physical Review B, 2019, 99, 014108
Effective site-energy model: A thermodynamic approach applied to size-mismatched alloys. F. Berthier, J. Creuze, B. Legrand, Physical Review B, 2017, 95, 224102
Ces études s'inscrivent dans un cadre vaste qui consiste à caractériser l’influence de l’environnement (principalement l’atmosphère mais on peut également penser à d’autres facteurs : substrat, matrice dans le cas de nanoprécipités ou de nanomatériaux composites, ...) sur la répartition des éléments au sein des nanoalliages. En effet, si les propriétés physiques particulières de ces objets sont souvent obtenues dans les conditions du vide, l’intégration technologique de ces matériaux est quant à elle assujettie à notre capacité à les protéger vis-à-vis de l’environnement ambiant, en particulier du dioxygène, sans perte de ces propriétés. Au contraire, dans le domaine de la catalyse hétérogène, la température et les pressions partielles des différents gaz sont les facteurs déterminants du point de vue de l’efficacité catalytique. Très souvent, l’état actif du catalyseur est atteint uniquement après un temps d’incubation, de quelques millisecondes jusqu’à plusieurs jours, l’existence d’un tel temps reflétant des modifications majeures au sein du catalyseur. Un exemple simple est celui de la formation d’un film proche de l’oxyde à la surface du catalyseur dans les réactions catalytiques d’oxydation. Enfin, la ségrégation superficielle induite dans les alliages métalliques en conditions de réaction, et donc les variations locales de structure et de composition, est un phénomène prédit et observé pour un certain nombre de systèmes bimétalliques. Ainsi, alors qu’une certaine configuration (structure, composition) peut présenter la propriété désirée, il est important de comprendre si cette configuration particulière est stable dans l’environnement d’utilisation pour une application spécifique. Un des exemples les plus standard est à nouveau probablement l’interaction entre le dioxygène et les surfaces métalliques, à la fois du point de vue (bon) de la catalyse et (mauvais) de la corrosion. Malgré leur importance, les détails concernant les mécanismes impliqués dans ces phénomènes sont encore très mal compris... et ne demandent qu’à l’être mieux !
Equilibrium Au-Pd(100) Surface Structures Under CO Pressure : Energetic Stabilities and Phase Diagrams. I. C. Oguz, T. Mineva, J. Creuze, H. Guesmi, The Journal of Physical Chemistry C, 2018, 122, 18922-18932
CO Adsorption-Induced Surface Segregation and Formation of Pd Chains on AuPd(100) Alloy: DFT-Based Ising Model and Monte Carlo Simulations. B. Zhu, J. Creuze, C. Mottet, B. Legrand, H. Guesmi, The Journal of Physical Chemistry C, 2016, 120, 350
Evidence of Pd segregation and stabilization at edges of AuPd nanoclusters in the presence of CO: a combined DFT and DRIFT study. B. Zhu, G. Thrimurthulu, L. Delannoy, C. Louis, C. Mottet, J. Creuze, B. Legrand, H. Gusemi, Journal of Catalysis, 2013, 308, 272
Ces études concernent les dépôts métalliques hétéroépitaxiés, i.e. métal A sur un substrat métallique B, qui sont généralement caractérisés par la formation de surstructures de surface et/ou par des phénomènes d’auto-organisation pour des taux de couverture submonocouches. L’auto-organisation de systèmes nanométriques a été étudiée intensivement ces dernières années en vue de leur utilisation potentielle pour la fabrication de nano-objets présentant des propriétés physiques ou catalytiques remarquables. Parmi ces systèmes auto-organisés, les surfaces vicinales et/ou facettées représentent une voie prometteuse pour la réalisation de substrats nanostructurés. Dans ce contexte et en collaboration avec Yves Garreau (Matériaux et Phénomènes Quantique (Université Paris 7/SOLEIL) et Alessandro Coati (SOLEIL), nous menons une étude sur l’évolution de la morphologie des surfaces vicinales de cuivre et de nickel induite par un dépôt d’argent en couplant des mesures obtenues par diffraction de rayons X en incidence rasante d’une part et des observations en microscopie à effet tunnel d’autre part avec des simulations numériques à l’échelle atomique. Ce couplage permet ainsi une meilleure détermination des reconstructions de surface et de mieux comprendre les forces motrices du facettage induit par adsorption.
Ag on a Ni vicinal surface: Coupling Stranski-Krastanov and “magic” heteroepitaxial growth. A. Bellec, Y. Garreau, J. Creuze, A. Vlad, F. Picca, M. Sauvage-Simkin, A. Coati, Physical Review B, 2017, 96, 085414
Incoherent Ag islands growth on Ni(100). J.-B. Marie, I. Braems, A. Bellec, C. Chacon, J. Creuze, Y. Girard, S. Gueddani, J. Lagoute, V. Repain, S. Rousset, Surface Science, 2017, 656, 101-108
AuNi alloy monolayer films electrodeposited on Au(111): An in situ STM study. F. Lecadre, F. Maroun, I. Braems, F. Berthier, C. Goyhenex, P. Allongue, Surface Science, 2013, 607, 25-32
Les nanoalliages peuvent apparaître dans des configurations très éloignées de celles d'équilibre. Ces configurations sont susceptibles d'évoluer vers les configurations d'équilibre. Pour étudier les cinétiques dans les nanoalliages, nous avons considéré un formalisme sur réseau, dont les paramètres ont été ajustés sur le système Cu-Ag. Deux types d'algorithme sont utilisés : l'un est stochastique (Monte Carlo Cinétique) et l'autre est déterministe, fondé sur le Champ Moyen Cinétique par site. Ces études sont menées en collaboration avec l'IRN Nanoalloys.
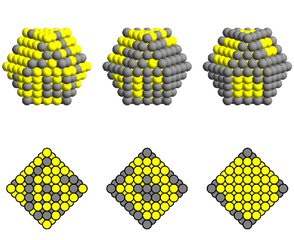
Évolution temporelle d'un cuboctaèdre CuAg : à gauche configuration initiale désordonnée, au centre configuration métastable de structure oignon, à droite configuration stable de type cœur-coquille.
Evolution temporelle du profil de concentration selon une coupe transversale : animateCubo11p6T900.avi
Proposition de stage/thèse sur le vieillissement des Nanoparticules AgPt (contact F. Berthier)
Ageing of out-of-equilibrium nanoalloys by a kinetic mean-field approach. F. Berthier, A. Tadjine, B. Legrand, Phys. Chem. Chem. Phys., 2015, 17, 28193-28199
Ces travaux concernent l’étude des modifications des comportements thermodynamiques de nanoparticules bimétalliques en fonction de leur taille et de leur morphologie, en comparaison avec les comportements observés en volume et en surface des alliages massifs (effet de taille finie sur les diagrammes de phase). De façon plus générale, il s’agit d’aboutir à une compréhension fine des moteurs pilotant les phénomènes de ségrégation superficielle à partir de la transposition de simulations numériques à l’échelle atomique (dynamique moléculaire ou simulations Monte Carlo utilisant des potentiels à N-corps ou des calculs ab initio) et de modèles déjà développés dans le cadre des alliages semi-infinis (méthodes de champ moyen ou Monte Carlo sur réseau rigide incluant les effets de structure de manière effective). Un des points clés réside dans la prise en compte du couplage entre réarrangements atomiques, locaux (relaxations) ou collectifs (changement de structure de la nanoparticule), et chimiques (ordre, ségrégation, désordre), couplage qui constitue une des difficultés inhérentes aux systèmes de petites tailles et qui peut bouleverser l’un ou l’autre des phénomènes précités.
Direct measurement of the surface energy of bimetallic nanoparticles: Evidence of Vegard’s rule-like dependence. A. Chmielewski, J. Nelayah, H. Amara, J. Creuze, D. Alloyeau, G. Wang, C. Ricolleau, Physical Review Letters, 2018, 120, 025901
Magic compositions in Pd-Au nanoalloys. B. Zhu, A. Front, H. Guesmi, J. Creuze, B. Legrand, C. Mottet, Computational and Theoretical Chemistry, 2017, 1107, 49-56
Stability Diagram of Janus and Core–Shell Configurations in Bimetallic Nanowires. E. Maras, F. Berthier, B. Legrand, J. Phys. Chem. C, 2016, 120, 22670-22680
Crossover among structural motifs in Pd-Au nanoalloys. B. Zhu, H. Guesmi, J. Creuze, B. Legrand, C. Mottet, Physical Chemistry Chemical Physics, 2015, 17, 28129
Phase diagrams of nanoalloys: influence of size and morphology. F. Berthier, E. Maras, B. Legrand, Phys. Chem. Chem. Phys., 2015, 17, 28347-28353